





Emai:marketing@yakkaa.com
業務谘詢專線:400-780-8018
Tel: +1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email: marketing@medicilon.com
地址:上海市浦東新區川大路585號
郵編:201299
電話:+86 (21) 5859-1500(總機)
傳真:+86 (21) 5859-6369







© 2023 上海hjc黄金城生物醫藥股份有限公司 保留所有權利 滬ICP備10216606號-3
滬公網安備 31011502018888號 | 網站地圖











業務谘詢
中國:
Email: marketing@yakkaa.com
業務谘詢專線:400-780-8018
(僅限服務谘詢,其他事宜請撥打川沙總部電話)
川沙總部電話: +86 (21) 5859-1500
海外:
+1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email:marketing@medicilon.com




一、蛋白質表達與純化實驗原理
(一)利用大腸杆菌表達進行原核表達
基因工程的最終目的是在一個合適的係統中,使外源基因高效表達,從而生產有價值的蛋白質產品。
1.原核生物基因表達的特點
(1)原核生物(如大腸杆菌)隻有一種RNA聚合酶,識別原核的啟動子,能夠催化所有RNA合成。
(2)原核生物的基因表達是以操縱子為單位。操縱子是數個相關的結構基因及其調控區的結合,是一個基因表達的協同單位。
(3)由於原核生物無核膜,所以轉錄與翻譯是耦聯的,二者也是連續進行的。原核生物染色體DNA是裸露的環型DNA,轉錄成mRNA後,可直接在胞漿中與核糖體結合翻譯成蛋白質。每個核糖體可獨立完成一條鏈的合成,多核糖體可同時在一條mRNA合成多條肽鏈,大大提高了翻譯效率。
(4)原核基因不含內含子(intron),沒有轉錄後加工過程。
(5)原核生物基因表達的控製主要是在轉錄水平。對RNA合成的調控有兩種方式:啟始控製(啟動子控製)和終止控製(衰減子控製)。
(6)在大腸杆菌mRNA的核糖體結合位點上,有一個轉譯啟始密碼子及同16S核糖體RNA 3′末端堿基互補的序列,即SD序列,真核基因則無此序列。
2.外源基因在原核中表達的關鍵
(1)表達載體將外源基因導入宿主菌,指導宿主菌的酶係統合成外源蛋白;
(2)外源基因不能帶有內含子;
(3)利用原核細胞的強啟動子和SD序列等控製外源基因的表達;
(4)外援基因與表達載體連接後,必須形成正確的開放閱讀框架;
(5)利用宿主菌的調控係統,調節外源基因的表達,防止表達的外源基因產物對宿主菌的毒害。
3.外源基因在原核細胞中表達的重要調控元件
(1)啟動子
-35區:位於轉錄啟始位點上遊35bp處,一般由10bp組成,與RNA聚合酶δ亞基結合;
-10區(TATA box):位於轉錄啟始位點上遊5-10bp處,一般由6-8bp,富含A T,與RNA聚合酶的核心酶結合。
原核表達載體所用的啟動子必須是原核啟動子,通常所用的可調控的強啟動子有:lac(乳糖啟動子)、trp(色氨酸啟動子),λPL(λ噬菌體左向啟動子),Tac(乳糖和色氨酸的雜和啟動子)等。
(2)SD順序
mRNA在細菌中轉譯率嚴格依賴於SD序列以及SD序列與啟始密碼子AUG之間的距離,例如,當lac啟動子的SD序列距AUG為7個核苷酸時,IL-2表達最高,為2581單位,而間隔8個核苷酸時,表達水平降到不足5個單位。另外某些蛋白質與SD序列結合也回影響蛋白質的翻譯。
(3)終止子(terminator)
4.原核表達載體的類型
(1)非融合型
不與細菌的任何蛋白或多肽融合在一起的表達蛋白。
優點:非常接近於生物體內天然蛋白質;
缺點:容易被細菌蛋白酶破壞,未知蛋白難於純化。
(2)融合型
蛋白質的一端由原核DNA序列或其它序列編碼,另一端由真核DNA的完整序列編碼。
優點:應該避免細菌蛋白酶的破壞,由於融合部分的關係,常易於表達產物的分離純化。
缺點:由於細菌的一段蛋白或多肽的存在,有時會影響其結構,需去掉。
5.用於原核細胞表達的外源基因
由於原核細胞缺乏真核細胞轉錄後的加工係統,mRNA的內含子不能切除,成熟的mRNA不能形成,同時原核細胞也缺乏真核細胞翻譯後的加工係統。隻能用cDNA而不能用基因組DNA,或體外合成基因,PCR擴增基因等。
(二)利用HIC樹脂純化ZsGreen蛋白
甲基化疏水反應層析(HIC)樹脂是利用疏水基團進行分離蛋白的一種簡單而有效的方法。結合緩衝液中的Cl-排斥帶負電荷的ZsGreen分子的β-外殼,這種排斥使分子內部外翻,露出疏水基團。露出的疏水基團牢牢與HIC樹脂的非極性甲基基團結合;中性鹽洗脫使ZsGreen保持為疏水狀態;但是可以洗脫未結合或者是結合弱的蛋白;低鹽緩衝液使疏水基團回到ZsGreen分子的原來位置,從而使ZsGreen分子HIC樹脂中釋放。
(三)SDS-PAGE分析
聚丙烯酰胺凝膠電泳是網狀結構,具有分子篩效應,它有兩種形式,一種是非變性聚丙烯酰胺凝膠,蛋白質在電泳中保持完整的狀態,蛋白在其中依三種因素分開:蛋白大小,形狀和電荷。而SDS-PAGE僅根據蛋白分子量亞基的不同而分離蛋白。這個技術首先是1967年由shapiro建立,他們發現在樣品介質和丙烯酰胺凝膠中加入離子去汙劑和強還原劑後,蛋白質亞基的電泳遷移率主要取決於亞基分子量的大小,電荷因素可以忽視。
SDS是陰離子去汙劑,作為變性劑和助溶試劑,它能斷裂分子內和分子間的氫鍵,使分子去折疊,破壞蛋白分子的二、三級結構。而強還原劑如巰基乙醇,二硫蘇糖醇能使絆胱氨酸殘基間的二硫鍵斷裂。在樣品和凝膠中加入還原劑和SDS後,分子被解聚成多肽鏈,解聚後的氨基酸側鏈和SDS結合成蛋白——SDS膠束,所帶的負電荷大大超過了蛋白原有的蛋白量,這樣就消除了不同分子間的電荷差異和結構差異。SDS-PAGE一般采用的是不連續緩衝係統,於連續緩衝係統相比,能夠有較高的分辨率。
濃縮膠的作用是有堆積作用,凝膠濃度較小,孔徑較大,把較稀的樣品加在濃縮膠上,經過大孔徑凝膠的遷移作用而被濃縮至一個狹窄的區帶。當樣品液和濃縮膠選Tris-HCl緩衝液,電極液選Tris-甘氨酸。電泳開始後,HCl解離成Cl-,甘氨酸解離出少量的甘氨酸根離子。蛋白質帶負電荷,因此一起向正極移動,其中Cl-最快,甘氨酸根離子最慢,蛋白居中。電泳開始時Cl-泳動率最大,超過蛋白,因此在後麵形成低電導區,而電場強度與低電導區成反比,因而產生較高的電場強度,使蛋白和甘氨酸根離子迅速移動,形成以穩定的界麵,使蛋白聚集在移動界麵附近,濃縮成一中間層。
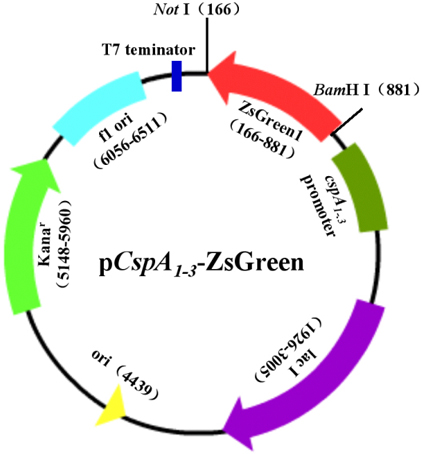
本實驗中,我們首先提取大腸杆菌(E.coli)基因組,PCR擴增冷激蛋白CspA的啟動子、上遊調控及下遊5′-UTR區序列,得到cspA1、cspA2和cspA3,在兩端引入酶切位點;用限製性內切酶切割質粒pZsGreen1-1,得到綠色熒光蛋白ZsGreen報告基因;以pET-28a(+)為骨架,酶切、連接,構建含冷激啟動子、報告基因為ZsGreen的表達載體pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen(圖1)及對照常溫表達載體pT7-ZsGreen(圖5-1)。
二、實驗儀器及藥品 (1)STE的配製(表3-1)
表3-1STE(100mL)配方
| 試劑名稱 | 試劑等級 | 用量 | 最終濃度 |
5mol/L NaCl | 分析純 | 2mL | 0.1mol/L |
1mol/L Tris-HCl(pH8.0) | 分析純 | 1mL | 10mmol/L |
0.5mol/L EDTA(pH8.0) | 分析純 | 200μL | 1mmol/L |
dH2O | 不需滅菌 | up to 100mL | — |
在60mL dH2O中加入5mol/L NaCl 2mL、1mol/L Tris-HCl(pH8.0) 1mL和0.5mol/L EDTA(pH8.0)200μL,混勻,加dH2O定容至100mL,1.034×105Pa高壓蒸汽滅菌15min,備用。配製好的STE含有0.1mol/L NaCl,10mmol/L Tris-HCl和1mmol/L EDTA。
(2)溶液I的配製(表3-2)
表3-2溶液I(100mL)配方
| 試劑名稱 | 試劑等級 | 用量 | 最終濃度 |
葡萄糖 | 分析純 | 0.9g | 50mmol/L |
1mol/L Tris-HCl(pH8.0) | 分析純 | 2.5mL | 25mmol/L |
0.5mol/L EDTA(pH8.0) | 分析純 | 2mL | 10mmol/L |
dH2O | 不需滅菌 | up to 100mL | — |
60mL dH2O中加入0.9g葡萄糖、1mol/L Tris-HCl(pH8.0) 2.5mL和0.5mol/L EDTA(pH8.0) 2mL,混勻,加dH2O定容至100mL,1.034×105Pa高壓蒸汽滅菌15min,4℃貯存。配製好的溶液I含有50mmol/L葡萄糖,25mmol/L Tris-HCl和10mmol/L EDTA。
(3)溶液II的配製(表3-3)
溶液II中含有0.4mol/L NaOH和1% SDS,該試劑需要新鮮配製,配製後將NaOH和SDS按1:1的比例混合。
表3-3溶液II(1mL)配方
| 試劑名稱 | 試劑等級 | 用量 | 最終濃度 |
5mol/L NaOH | 分析純 | 40μL | 0.4mol/L |
dH2O | 不需滅菌 | up to 500μL | — |
2%SDS | 分析純 | 500μL | 1% |
dH2O | 不需滅菌 | up to 500μL | — |
(4)溶液III的配製(表3-4)
表3-4溶液III的配製(100mL)
| 藥品名稱 | 用量 |
5mol/L KAc | 60mL |
冰醋酸 | 11.5mL |
dH2O | 28.5mL |
配製好的溶液III含3mol/L KAc、5mol/L冰醋酸(pH4.8)。
三、質粒抽提實驗方法
質粒提取過程如下:
(1)將瓊脂培養板上的陽性單菌落,移至3mL LB液體培養基中(含Amp或Kana),或將大腸杆菌的甘油菌按1:30的比例接到LB液體培養基中,37℃劇烈搖蕩培養過夜;
(2)取500μL菌體,與40%甘油等體積混合,-70℃保存,剩餘的菌體用於質粒的提取;
(3)取出1.5-2mL菌液移至Eppendorf管中,12000rpm,4℃離心30s,棄上清,用1mL STE懸浮菌體沉澱,洗滌菌體,再離心回收菌體;
(4)再重複用STE漂洗菌體,離心後,去盡上清液;
(5)將細菌沉澱懸浮於100μL冰預冷的溶液I中,強烈振蕩混勻;
(6)加入200μL新配製的溶液II,輕輕顛倒離心管5次以混合內容物,不要強烈振蕩,放置冰上3min左右(根據不同菌株,可適當縮短);
(7)加入150μL冰預冷的溶液III,輕輕顛倒5次,混勻,置於冰上5min;
(8)15000rpm,4℃離心5min,將上清轉移到另一個離心管中;
(9)向上清中加入等體積的酚:氯仿(1:1),混勻,15000rpm離心5min,將上清轉移到另一個離心管中;
(10)加入2倍體積冰預冷的無水乙醇,室溫放置2min,沉澱雙鏈DNA;
(11)15000rpm,4℃離心5min;
(12)倒掉上清,使液體盡量流盡,空氣中幹燥沉澱;
(13)用20μL含有RNaseA的TE緩衝液溶解核酸沉澱,瞬時離心,混勻,貯存於-20℃冰箱中備用。
四、實驗結果
(一)未純化SDS-PAGE分析
將含有pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen的大腸杆菌(E.coli),在16℃表達;含有pT7-ZsGreen的大腸杆菌(E.coli)在37℃下表達至OD600=0.6時,加入IPTG誘導表達,不加IPTG誘導的作為對照。誘導表達5h後收集菌體,進行SDS-PAGE分析(圖5-2)。
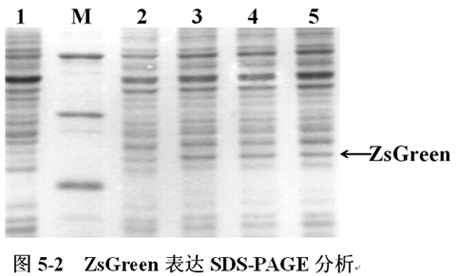
圖5-2中,M為低分子量蛋白Maker。1-5分別為pT7-ZsGreen(IPTG-,37℃)、pT7-ZsGreen(IPTG+,37℃)、pCspA1-ZsGreen(16℃)、pCspA2-ZsGreen(16℃)、pCspA3-ZsGreen(16℃)表達ZsGreen。箭頭所示為ZsGreen,其大小約為27KD。SDS-PAGE結果顯示:三種低溫誘導表達載體pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen在16℃下表達ZsGreen正常,可用作下一步對其翻譯效率的分析。
(二)純化後SDS-PAGE分析
將含有低溫誘導表達載體pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen的大腸杆菌(E.coli),在16℃下表達,以1h為時間間隔收集菌體,利用HIC樹脂純化ZsGreen蛋白,進行SDS-PAGE分析(圖5-3)。
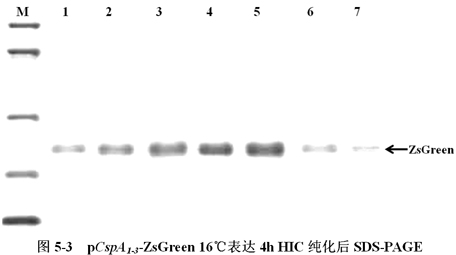
圖5-3中,M為低分子量蛋白Maker。A1-7、B8-14、C15-21分別為pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen 16℃表達4h HIC純化後SDS-PAGE。箭頭所示為ZsGreen,其大小約為27KD
 相關新聞
相關新聞川沙總部
地址: 上海市浦東新區川大路585號
郵編: 201299
電話: +86 (21) 5859-1500(總機)
傳真: +86 (21) 5859-6369
海外:
Email: marketing@medicilon.com
Tel: +1(626)986-9880(U.S. - West Coast)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)


 關於我們
關於我們










