2017年依然是藥品製造業研發生產政策發布的大年。
1月10日,CFDA藥品審評中心發布了關於《已上市化學藥品生產工藝變更研究技術指導原則》征求意見的通知(以下簡稱“2017年版指導原則”)。這是自2008年原國家食品藥品監督管理局發布的《已上市化學藥品變更研究的技術指導原則(一)》(以下簡稱“2008年版指導原則”)以來的首次調整。
“2017年版指導原則”主要按原料生產和製劑生產角度進行生產工藝變更研究,並重點以原料藥、口服固體製劑和注射劑3種市麵上化學藥品的主流劑型製定變更研究及信息要求。隨著“2017年版指導原則”的發布,2016年8月CFDA發起的藥品生產工藝核對工作將會在上述3個劑型優先開展。
兩大亮點
一與國際現行標準接軌
“2017年版指導原則”非常注重與國際現行標準接軌,表現在3個方麵:
第一,仿製藥生產工藝變更後,強調與原研產品的等同性或等效性,評估方法需符合國際現行標準。
第二,多次參考國際通行指導原則,如原料藥中新雜質的控製參考ICH Q3規定的質控限度。參考文獻也涵括了FDA、EMEA、TGA和ICH有關指導原則。
第三,注重產品在國內外的藥典收載情況,並要求對原批準質量標準、現行版中國藥典標準以及現行版國外主流藥典標準進行比較。
縱深
“2017年版指導原則”要求生產工藝變更後的質量要原研產品一致,並且評估方法要與國際現行標準接軌,這與國家目前推行仿製藥一致性評價政策方向相一致。結合工藝核對工作,工藝變更的研究將倒逼口服固體製劑和注射劑開展與原研產品質量一致、臨床等效的研究。
按照工藝核對的時間表,藥品生產企業應於2017年6月30日前完成在產品種生產工藝的研究驗證、提交補充申請等相關工作,其他暫不生產品種應於2017年12月31日前完成上述工作;未按時完成的,應停止生產。
“2017年版指導原則”增加了與原研一致的要求,預計會有部分產品因此而停產。不在首批必須在2018年完成的289個一致性評價產品目錄的產品應積極開始一致性評價了。
二更注重流程管控
⒈重點參考2008版三個項目
“2008年版指導原則”將工藝變更研究分為原料藥生產工藝變更、藥品製劑處方中已有藥用要求的輔料和製備工藝變更、注冊標準變更、規格變更、有效期和貯藏條件變更、藥品的包裝材料和容器變更、進口藥品產地變更、進口原料藥產地和進口藥品所用原料藥產地變更、變更國內生產藥品製劑的原料藥產地10個項目。
2016年8月發布的關於開展藥品生產工藝核對工作的公告,對Ⅰ類變更、Ⅱ類變更和Ⅲ類變更的劃分更多是參考“2008年版指導原則”的“變更原料藥的生產工藝”和“藥品製劑變更製劑的生產工藝”,其餘項目基本沒有提及。
“2017年版指導原則”除了參考變更“原料藥的生產工藝”和“藥品製劑變更製劑的生產工藝”,還參考了“2008年版指導原則”中“變更藥品製劑處方中已有藥用要求的輔料”,並整合在“變更藥品製劑生產工藝”中。
2增加工藝流程關鍵環節點的劃分
“2008年版指導原則”是在十大項目下劃分Ⅰ類變更、Ⅱ類變更和Ⅲ類變更層次對應的工藝變更細節內容。
“2017年版指導原則”則是在變更原料藥生產工藝和變更藥品製劑生產工藝兩大項目下增加對工藝變更流程關鍵環節點的劃分,然後在關鍵環節點上細分微小變更(即Ⅰ類變更)、中等變更(即Ⅱ類變更)和重大變更(即Ⅲ類變更)對應的不同層次的細節內容。
其中,變更原料藥生產工藝就包括變更生產路線(如縮短、延長或調整生產路線,變更試劑和起始原料等)、變更生產條件(如攪拌方式、幹燥方式等工藝原理變更,投料量、反應溫度、反應時間、攪拌速度、攪拌時間等工藝參數變更)、變更物料控製/過程控製(如變更試劑、起始原料來源、製備工藝、質量控製等)及其它可能的變更。
化學藥品生產工藝變更主要包括變更輔料(來源、型號、級別、用量、種類等)、變更生產設備、變更製備工藝(工藝原理變更如幹法製粒和濕法製粒的互變,工藝條件變更如幹燥溫度、壓片硬度等)、變更製劑生產過程質量控製方法及其限度(中間體質量標準變更、過程檢驗項目變更)等。
增加對工藝變更流程關鍵環節點的劃分,讓“2017年版指導原則”更貼近製藥工程學的思維。
3三層次所歸類的細節內容更具體化
增加“對工藝變更流程關鍵環節點”這一個維度後,“2017年版指導原則”結合微小變更、中等變更和重大變更三層次分析不同細節內容的影響程度,更能具體化不同工藝變更的細節內容在工藝變更中能引起哪種層次的影響,從而達到研究驗證的效果。
例如在“2008年版指導原則”中,輔料用量變更是放在Ⅱ類變更的,而在“2017年版指導原則”中,輔料用量變更卻有三層次的變更,而這三層次的變更程度與劑型有相關性(見表1、2、3)。
表1 Ⅰ類變更(即微小變更)對比
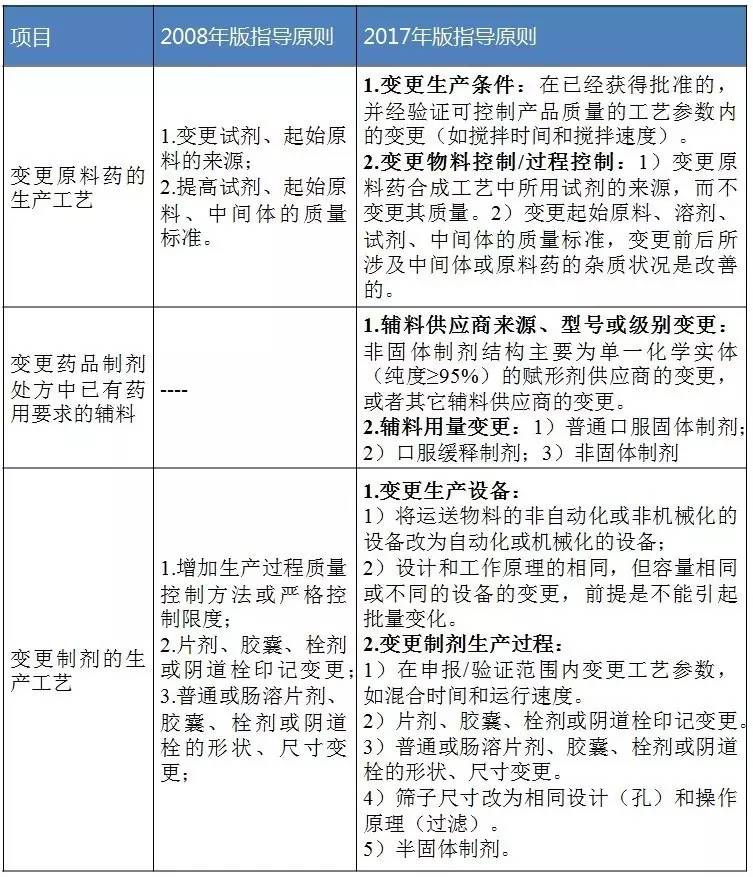
數據來源:識敏信息(注:“關於開展藥品生產工藝核對工作的公告”相關內容參考“2008年版指導原則”,但輔料除外。“2017年版指導原則”將輔料變更內容放在“變更藥品製劑生產工藝”部分。下同。)
表2 Ⅱ類變更(即中度變更)對比
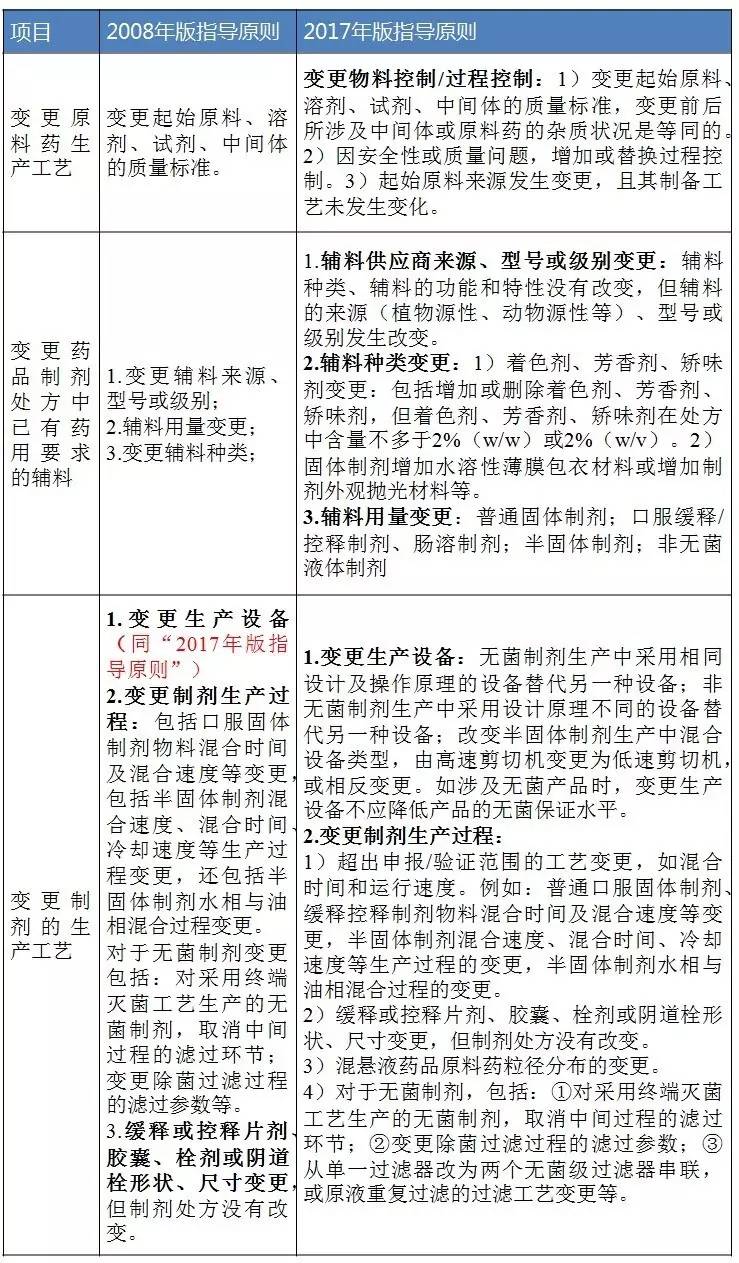
數據來源:識敏信息
表3 Ⅲ類變更(即重大變更)對比
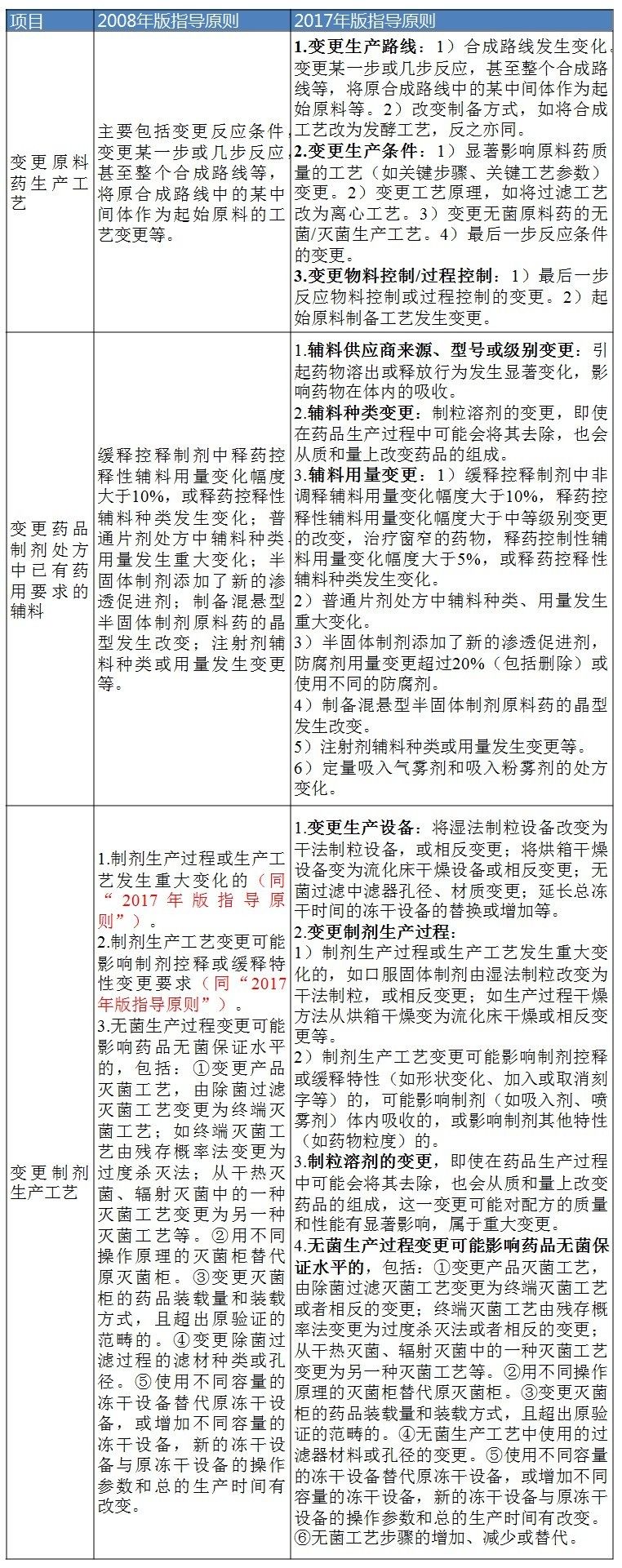
數據來源:識敏信息
輔料用量變更的微小變更:是指對產品質量和性能不產生任何影響。變更前後藥物溶出/釋放行為保持一致,或與體內吸收和療效有關的重要理化性質和指標保持一致。除產品外形外,變更後藥品質量標準沒有改變或更加嚴格。
輔料用量變更的中等變更:是指變更前後藥物溶出/釋放行為保持一致,或與體內吸收和療效有關的重要理化性質和指標保持一致。除產品外形外,變更後藥品質量標準沒有改變或更加嚴格。
輔料用量變更的重大變更:是指對藥品質量可能產生較顯著的影響。如半固體製劑添加了新的滲透促進劑;半固體製劑防腐劑用量變更超過20%(包括刪除)或使用不同的防腐劑。製備混懸型半固體製劑原料藥的晶型發生改變。
縱深
“2017年版指導原則”更加全麵在從工藝設計、工藝流程管理角度去研究驗證生產工藝變化對藥品的安全性、有效性和質量可控性的影響。企業在學習“2017年版指導原則”時,應同時領會CFDA對於工藝管理從碎片化到係統化的改變。
未來展望
1.預計《關於開展藥品生產工藝核對工作的公告》所提及的中藥和生物製品的指導原則也將要發布。
2.“2017年指導原則”嚴謹度較2008年版提高,體現了國家提升仿製藥藥品質量的決心,為化學藥生產企業豎了一個新的門檻。工藝核對的飛行檢查將會曝光一些不合規生產的企業,工藝核對將成為生產企業新的洗牌大殺器。
3 .化學藥企業應積極提升競爭力,往國際標準靠攏,爭取在優勝劣汰競爭中“剩者為王”。




























 相關新聞
相關新聞

 關於我們
關於我們










