首仿藥的概念,最早來自美國,起源於1984年9月24日頒布的藥品價格競爭與專利期補償法案(Drug Price Competition and Patent Term Restoration Act,又稱Hatch-Waxman法案)。
該法案規定,在“專利無效或者批準正在申請的藥物不會侵犯專利”的情況下,第一個向美國食品藥品管理局(FDA)遞交簡化新藥申請(Abbreviated New Drugapplication,簡稱ANDA)的申請者將擁有180天的市場獨占期。在這180天內,仿製藥企業可以快速收回投資,並在市場被其他仿製藥充斥之前確立其地位。因此,自Hatch-Waxman法案頒布以後,越來越多的國內外醫藥企業加入了首仿藥的爭奪戰中。
中國作為仿製藥大國,近年來也開始關注美國首仿藥,許多醫藥名企在推進醫藥國際化的進程中,都已經紛紛開始研發和申報美國首仿藥。
一方麵,隨著中國藥企技術創新水平的提高和高端人才的引進,中國生產的仿製藥已經可以達到歐美製藥標準,這無疑給開發美國首仿藥提供了一個良好的基礎。另外一方麵,國內藥企擁有多年的原料藥生產和出口經驗,因此從技術層麵上講,中國已經具備申報美國首仿藥的基礎和能力。
除了技術平台,國內知名藥企還配備了較為完備的法律法規平台、藥品國際注冊平台、專利平台和臨床試驗平台。尤其近幾年以來,國內製藥行業產業鏈發展日趨成熟和完備,眾多的新藥開發CRO(包括臨床前CRO、臨床CRO、注冊CRO等等)如雨後春筍般嶄露頭角,為醫藥研發及國際化提供了全新的產業發展生態環境。
本文通過分析Hatch-Waxman法案和藥品專利鏈接製度、首仿藥製度,以期為國內企業申報首仿藥,以及美國首仿藥製度對中國藥企的啟示。
1、Hatch-Waxman法案的產生和發展
昂貴的藥品費用對消費者構成沉重負擔,嚴重影響著公眾的健康。原研藥價格居高不下,是由於專利權人須按照FDCA法案(Food,Drug and Cosmetics Act)規定,通過大量的臨床試驗以證明原研藥品的有效性及安全性,這個過程不僅需要消耗大量資金而且耗時漫長,通常在專利期過去一大半時才能完成FDA的審批。對於仿製藥,價格雖低於原研藥,但FDA規定了與原研藥同樣嚴格的申請程序,這也極大的消減了仿製藥企業的積極性,束縛了仿製藥行業的發展。
羅氏(Roche)公司訴博拉(Bolar)公司一案,促使了Hatch-Waxman法案的產生,有效地解決了以上問題。1983年,Bolar公司開發了Roche公司原創的鎮靜催眠藥——鹽酸氟西泮的仿製藥,並希望可以盡快上市,其於該產品的專利期屆滿前,從加拿大進口鹽酸氟西泮原料藥進行試驗,以收集FDA審批所需的生物等效性試驗等數據。Roche公司認為Bolar公司的行為侵犯了其專利權,遂將其訴至法院。美國紐約東區地方法院認為Bolar公司的目的是為了進行試驗,以“試驗使用例外”判定Bolar公司不構成侵權。Roche公司不服,提起上訴,經過雙方爭辯,聯邦巡回上訴法院最終裁定Bolar公司為藥品準備上市進行生物等效性試驗是有商業目的的,不適用“試驗使用例外”,因此構成侵權。盡管Bolar公司敗訴,但其在上訴程序中提出的“不合理延長了專利保護的期限”的抗辯理由引起了美國國會的重視[3]。1984年,由美國國會議員森納托·哈奇(Senator Hatch)和亨利·瓦克斯曼(Henry Waxman)聯合提議,並經國會批準,發布了Hatch-Waxman法案。該法案第202條規定,在美國製造、使用或銷售藥品,需要依照聯邦藥品管理法的規定提交相關研發信息,僅僅為滿足聯邦法律對提交數據的規定而進行的相關行為,如在美國本土製造、使用、許諾銷售或銷售專利藥品或將專利藥品進口至美國本土不認為是專利侵權行為。這就是著名的“Bolar例外”條例,該條例後來被編入美國法典35 U.S.C §271(e)(1),也就是美國專利法的第156條,稱為“避風港條款”。除“Bolar例外”條例外,Hatch-Waxman法案還包括ANDA、延長專利製度、鼓勵專利挑戰等內容。
Hatch-Waxman法案對原研藥企業進行了有效補償,解決了原研藥專利權人因FDA審批未完成,而使得專利授權後仍無法上市,導致原研藥保護期限受損的問題。更重要的,該法案鼓勵了仿製藥企業的發展,解決了仿製藥在原研藥專利權到期後無法及時上市的問題。Hatch-Waxman法案有效地平衡了原研藥企業和仿製藥企業的利益,同時也平衡了原研藥企業和公眾健康之間的利益,是確立美國藥品專利鏈接製度(Pharmaceutical Patent LinkageSystem)的基礎。之後,為了解決製度中存在的問題,美國國會在2003年又相繼通過了《醫療保險處方藥改良和現代化法案》(Medicare Prescription Drug Improvement andModernization Act,MMA)和《更容易獲得可支付藥品法》(Greater Access to Affordable Pharmaceutical Act,GAAP),修改了與專利鏈接相關的FDA工作程序、法院訴訟程序和限製競爭等方麵的規定。
2、美國藥品專利鏈接製度(Pharmaceutical Patent Linkage System)
美國藥品專利鏈接是指仿製藥上市批準與新藥專利期滿相“鏈接”,即仿製藥注冊申請應當考慮先前已上市藥品的專利狀況,從而避免可能的專利侵權。美國藥品專利鏈接製度有兩層含義:一是仿製藥的上市申請審批與相應的藥品專利有效性審核的程序鏈接;二是美國FDA與美國專利商標局(USPTO)的職能鏈接。具體地,該製度包含以下5個方麵的內容:
(1)藥品專利期延長(Patent Term Extension,簡稱PTE)製度
專利期延長製度是對藥品和醫療器械因臨床試驗和行政審批所喪失的專利期予以補充的一種製度。眾所周知,新藥開發具有投資大、風險高、難度大和周期長的特點,從發現可能成藥的新的先導化合物,申請專利開始,要進行漫長的研究工作,才能最終把一個新藥推向市場。在生物醫藥領域,每一個重磅炸彈級新藥的推出,都代表著有一張嚴密的藥物專利保護網絡在默默守護創造者的智慧結晶,原研藥企業通過不斷推陳出新、持續技術創新與知識產權布局,便可牢牢把握市場主動權。但是,往往相當多的藥品上市後,核心專利可以保護的有效期限已經很短。為了調動原研藥企業的積極性,使其獲得足夠的藥物研發回報,Hatch-Waxman法案規定了新藥申請者可獲得專利延長期,補償其在臨床試驗和藥品審評中所消耗的時間,但最多不超過5年,並且延長期限加上藥物上市時所剩餘的專利期限之和不能超過14年。但是,如果是申請人未盡到應有注意義務而導致專利期限耽擱的,該期限不計入延長期內。對同一藥物隻能申請一次專利期間延長,即使該藥物有多項專利,而且僅藥物的核心專利可延長,後續專利不能延長。
原研藥企業通常會選擇將PTE加在其中一項化合物專利上,以保證其最核心的專利可以有效阻止仿製藥的上市,但是PTE的確定,有時卻是一個漫長的過程。因此仿製藥企業研發產品時,要格外留意,尤其是首仿藥的申報者,PTE延長直接決定著其專利策略的製定。
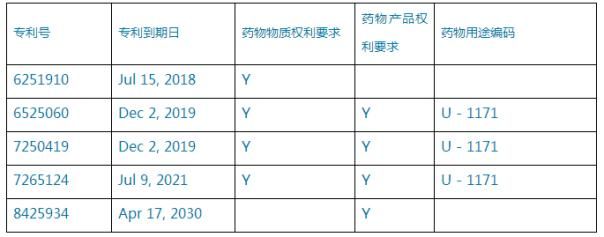
案例1:比如阿斯利康在美國上市的藥物替格瑞洛(Ticagrelor),其專利信息如表1所示:
表1、替格瑞洛美國專利信息
其中,專利’910、’060和’419都是其化合物專利,’124為晶型專利,’934為製劑專利。替格瑞洛首仿藥的申報時間為2015年7月20日,訴訟30個月遏製期的結束日為2019年1月20日。如果專利情況僅限於此,我們可能就’910專利直接提出第III段聲明(PIII),等待其專利自動到期。但是經過調查發現,阿斯利康申請了PTE,並且已經獲批,隻是因為專利再授權問題,PTE的公布被大大延遲。意欲申報首仿藥的仿製藥企業不可能等到PTE公布後再申報,因此紛紛對’910專利提起了第IV段聲明(PIV),即認為該專利無效或不可實施。可見,鼓勵創新藥開發的PTE製度對仿製藥企業的影響是非常大的。
(2)市場獨占期(Market Exclusivity)規定
除專利期延長製度之外,FDA還給予獲批藥品一定的市場獨占期,包括:新化學實體(New Chemical Entity,簡稱NCE)獨占期5年、孤兒藥獨占期(Orphan Drug Exclusivity,簡稱ODE)7年、兒科用藥(Pediatric Medication,簡稱PED)6個月、新劑型、新用途等3年,等等。在市場獨占期內,FDA不會批準仿製藥企業上市相應的仿製產品,因此即使在該期限內專利過期,原研藥企業仍然可以單獨占領市場。
市場獨占期是對原研藥企業的研發激勵,但是對仿製藥企業也有一定的提示作用,尤其是首仿藥。根據FDA法規規定,仿製藥最早可以在NCE-1日申報,即新藥獲批後的第5年的第1天,也即新藥上市後滿4年,比NCE時間少了1年,因此稱為NCE-1日。該時間通常被認為是首仿日。所有的仿製藥企業如果做好了充分的準備,都會在這一天爭相向FDA遞交自己的ANDA資料,一旦資料被受理,其首仿資格便會獲得確認。
(3)橙皮書(Orange Book,簡稱OB)製度
橙皮書即《經治療等同性評價標準的藥品》(Approved Drug Products withTherapeutic Equivalence Evaluation),由美國FDA出版,其詳細完整地列出了獲得批準的藥品,以及該藥品涉及的專利和獨占期信息。根據Hatch-Waxman法案規定,原研藥企業在向FDA遞交新藥上市許可申請(New Drug application,簡稱NDA)時,必須同時提供專利信息。當該新藥獲批後,對應的專利就會登記在橙皮書中,為日後仿製藥企業開發仿製藥、進行ANDA、或專利訴訟提供參考資料。可以被橙皮書收錄的專利包括直接指向藥品的專利,如化合物、產品、晶型等,也可以包括治療方法。一般情況下,代謝物、包裝和工藝專利是不予收錄的。
(4)ANDA製度
Hatch-Waxman法案通過以前,關於仿製藥和新藥的審批程序的規定並無差別,仿製藥被要求進行同新藥注冊一樣的安全性及有效性試驗,這使生產仿製藥幾乎無利可圖。該法案通過後,申請仿製藥上市,申請人在提交ANDA時,隻需要進行生物等效性試驗,即證明與參比製劑具有生物等效性,而無需提交其他數據,大大降低了仿製藥企業的成本,從而加快了仿製藥上市,減少了患者的醫療費用開支。自ANDA實施以來,美國的仿製藥產業發展很快,FDA收到的仿製藥申請不斷增多,批準上市的仿製藥數量和銷售額也是逐年上升,例如從2011年到2015年,美國仿製藥市場就從513億美元增長到了762億美元(出廠價,不包括回扣和折扣)。
(5)專利挑戰製度
根據Hatch-Waxman法案規定,仿製藥企業商在遞交ANDA時,必須依照橙皮書的規定,遞交以下四種聲明之一:
第I段聲明(PI):該藥品無專利;
第II段聲明(PII):該藥品有專利,但該專利已經失效;
第III段聲明(PIII):在相關專利失效前,不要求FDA批準該仿製藥;
第IV段聲明(PIV):與申請的仿製藥相關的專利是無效的(策略1)或者仿製藥不侵權(策略2)。
其中,第一個向美國FDA遞交ANDA、並含有PIV聲明的仿製藥申請者,如果專利挑戰成功,則FDA將給予180天的市場獨占期。在這180天內,FDA不再批準其他的ANDA持有人上市,而獲得市場獨占期的仿製藥企業可以以原研藥60%-90%的價格在市場上銷售,以彌補其在專利挑戰時消耗的訴訟費用,並快速地收回成本。例如Barr公司在2011年8月成功挑戰了Eli Lilly公司的Prozac專利並上市其仿製藥氟西汀,其在180天市場獨占期內就獲得了31億美元的銷售額。該製度大大激勵了仿製藥企業向原研藥企業挑戰專利的積極性,由於價格競爭的因素,是否獲得180天市場獨占期對收益可以產生5~10倍的區別。當然,有些仿製藥企業即使不是第一個申請人(First Filer),不能獲得180天獨占期,但仍會選擇在遞交ANDA時提出PIV挑戰,因為如果成功地規避了原創的OB專利,或是OB所列專利不可實施或無效,則ANDA持有人可以排除保護期較長的專利的幹擾,盡早獲得FDA的批準,提前上市。據FDA數據顯示,2004年1月1日至2011年6月10日,提出PIV聲明的ANDA申請達766件,對應的原研藥為556個,其中,Teva、Mylan、Apotex、Sandoz等世界大型仿製藥企業均是提出PIV挑戰的主力軍。
PIV聲明的ANDA是典型的仿製藥挑戰專利,PIV聲明即為挑戰書。含PIV專利挑戰的產品是可以在NCE-1(即NDA批準日期滿48個月後的第一天)遞交ANDA的,這一天通常被稱為“首仿日”,如果在這一天有多家仿製藥企業遞交ANDA,那麽在專利挑戰成功後,它們將共同擁有180天的市場獨占期。含PIV聲明的申請者在其ANDA被受理後的20個工作日內,應及時通知NDA持有人和專利權人,並提供仿製藥產品未侵權或原研藥企業專利無效的法律依據。如果NDA持有人和/或專利權人在45天內提起專利侵權訴訟,則30個月遏製期啟動,在這30個月內,FDA會繼續審查ANDA申請人的材料,材料合格的給予臨時批準,但不會批準其上市。如果遏製期內訴訟判決專利無效或產品不侵權,則在判決生效之日臨時批準轉為正式批準,允許上市並獲得180天市場獨占期。
3、 美國首仿藥開發的專利策略
相對於普通仿製藥,美國首仿藥的開發時間相對較長,投入的成本也相對較多,但考慮到180天市場獨占期的巨額回報,許多企業仍然願意加入到首仿藥的激烈爭奪之中。
(1)立項前調研
開發一個美國首仿藥,首先要做好充分的信息調研準備工作,然後結合本企業自身的條件,討論是否開展項目研究。立項前的調研工作涉及藥品法律法規信息、藥品注冊信息、臨床信息、市場分析、專利評估和實驗技術考量等方麵的內容。其中專利評估是最為重要的環節之一。以諾華(Novartis)公司在2010年9月上市的原研藥鹽酸芬戈莫德(Fingolimod Hydrochloric,商品名Gilenya)膠囊為例,在2011年時,橙皮書中與Gilenya相關的專利隻有一篇,即2019年2月18日到期的US5604229。另外,橙皮書中還記載了該藥的NCE市場獨占期,到期日為2015年9月21日。經過計算,若申報首仿藥,訴訟的30個月遏製期滿日為2018年3月21日,早於’229專利的專利到期日,因此當時便可以考慮對該專利提起PIV聲明。由於’229專利是化合物專利,無法規避,隻能選擇策略1。之後再由美國專業律師分析該專利的有效性,是否有可以爭議的問題點,以爭取首仿藥的可能性。
此外,在評估時,除了橙皮書專利,研發人員還需要提前考慮和分析未授權的其他專利。依然以芬戈莫德為例,在2011年時,與鹽酸芬戈莫德相關的晶型專利、製劑專利都處於審查階段,而治療方法專利雖然已經授權,卻還未被橙皮書收錄,這些專利都會成為日後產品上市的障礙。經過商討,技術人員認為開發鹽酸芬戈莫德的新晶型和新製劑存在一定的可能性,可以考慮規避策略來應對還未被橙皮書收錄的專利。
(2)策略調整和技術改進
隨著專利審查曆程的推進,仿製藥企業往往可以做出對自己有利的策略改變。例如鹽酸芬戈莫德,其鹽酸鹽晶型最終沒有被美國專利局授權,考慮到開發成本,仿製藥企業完全可以放棄開發新晶型,直接沿用原研藥企業的晶型,以減小後期“生物等效性試驗”的風險。然而,原研藥企業Novartis的製劑專利US8324283最終獲得了授權,並且被收錄到橙皮書中,該專利的在美國的到期日為2026年3月29日,遠遠晚於30個月遏製期滿的時間,由於前期已經考慮到規避專利的策略,隻要能成功研發出新的處方,則在ANDA申報時,完全可以提出PIV聲明,又因為是不侵權的論點,在專利訴訟中將會有較大的優勢。
(3)專利聲明的確定
在所有研發工作結束後,專利策略就基本成型了,一般會考慮兩個時間點,一是首仿藥提交日,另一個是專利遏製期滿日。在ANDA遞交日前的專利,直接提PII聲明。在遞交日和遏製期滿日之間的專利,可以考慮提PIII聲明。而那些晚於遏製期滿的專利,對於晶型和製劑專利,如果已經順利規避的,可根據自主研發的晶型或處方提交PIV不侵權聲明,如果未成功規避,但有無效可能性的,則可以提出PIV無效聲明。對
於治療方法專利,除前述方法外,還可以提交“Litter viii Statement”,俗稱小八條聲明,即聲稱所申報藥品不會用於該專利所包含適應症的治療,這樣也可以有效地規避原研藥企業的專利。不過,隨ANDA遞交的專利聲明中,若隻有小八條聲明,是不可以給予180天獨占期獎勵的,還必須含有第PIV段聲明。另外還需注意,提出小八條聲明時,藥品說明書中對應適應症的相關信息要一並刪除。
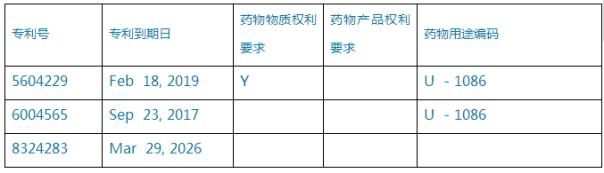
案例2:仍以鹽酸芬戈莫德為例,在2014年中準備ANDA申報時,其在橙皮書中的專利信息如表2所示:
表2、芬戈莫德美國專利信息
其中,’565專利為治療方法專利,其到期日早於30個月遏製期滿日(2018年3月21日),可直接提出PIII聲明;’229專利和’283專利,都晚於30個月遏製期滿日,可以同時提出PIV聲明。又因為’229專利為化合物專利,調研時已經分析過,可采取無效策略,而’283專利,在研發時已經規避了原研藥的處方,可直接提出不侵權。
綜上所述,對於美國首仿藥而言,專利策略貫穿於項目始終,且必須緊密結合美國專利鏈接製度和項目開發進程來不斷地進行調整,以期在向FDA遞交ANDA資料時呈上最完美的專利聲明,並為後期的專利訴訟創造最大的勝訴空間。
4、美國首仿藥製度對中國藥企的啟示
公開數據顯示,從2012年1月1日至2016年12月31日這5年間,全球將有631個專利藥到期,這一現象在業內被稱為“專利懸崖”[13]。原研藥的“專利懸崖”給仿製藥產業帶來的機遇已是行業共識,首仿藥更是成為“兵家必爭”之地。從國際經驗來看,仿製藥企業通過專利挑戰並取得美國首仿藥資格上市,不僅僅可為企業贏得巨額利潤,也通過技術和資金的積累,為企業下一步新藥研發創新提供重要的支撐與保障。因此,在當今醫藥市場多方博弈的競爭格局中,我國醫藥企業在美國提交ANDA申請,開展仿製藥專利挑戰,爭取180天獨占期,不僅僅能夠以較小的成本賺取比普通仿製藥以及國內仿製藥品更高額的利潤回報,獲得與輝瑞(Pfizer)、默克(Merck)、諾華(Novartis)等跨國藥業巨頭競爭的主動權,更能夠迅速提升我國醫藥企業的綜合實力,從而實現我國醫藥產業結構的全麵調整。許多國內企業如東陽光藥、華海藥業等已經走上了美國首仿藥爭奪的戰場。
中國仿製藥企業想要爭奪美國首仿藥市場,首先必須要深入了解與研究美國的相關藥品專利法律保護製度;其次要重點進行目標藥物的專利評估,根據實際的利益權衡,選擇合適的仿製藥申請途徑;再次需要企業提升自身及產品的品質。相信隨著中國藥企研發與技術創新能力的不斷提升、研發項目管理能力的不斷優化、藥物細分領域產品定位能力的不斷清晰,以及中國藥企實施“走出去”的國際化進程的決心,中國藥企完全可以慢慢拉近與國外大型仿製藥企業的距離,在美國首仿藥市場上分得一杯羹。




























 相關新聞
相關新聞

 關於我們
關於我們










