眾所周知,一個分子從實驗室到成為一個藥品,是一個漫長而艱辛的旅程。但是,對於這個旅程各個階段的作用和目標,行業內真正能夠全麵了解的卻並不多。
近日兩位專業的資深作者(諾華法規事務經理Sharry Arora、Six-Sigma公司項目管理專家Bhaskar Saxena)在《Regulatory Focus》發表題為“Journey of a Drug From Concept to Approval”一文,對新藥研究開發的過程和狀況做了比較全麵、簡要而清晰的介紹。本文基本保持原貌,筆者作少許補充,供相關人員參考。
縱觀從實驗室到新藥上市的整個過程,包括尋找先導化合物(藥物發現)、目標確定(藥物開發)、臨床前試驗、臨床試驗和後期生產研究的漫長曆程,篩選和評估數千個甚至數百萬個化合物,或許經曆10年或更長時間,最終隻有少數新藥獲得批準,在藥房貨架上獲得一席之地。
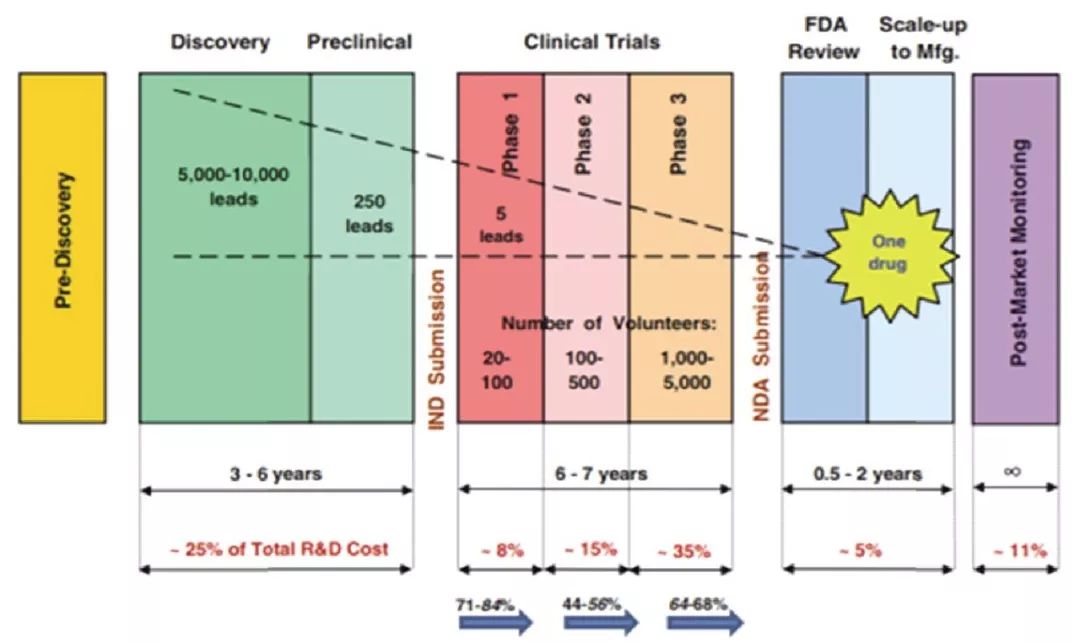
大數據亦可反映出新藥開發之艱難:僅臨床試驗就可能需要6~7年時間;研究和開發成功一種新藥平均成本約為26億美元(包括失敗造成的損失);進入臨床試驗的藥物最終被批準的可能性低於12%;5000個分子中隻有1個最終成為處方藥。
Step1 早期發現
階段目標 靶標的鑒別和驗證
新藥的最早期階段,應該追溯到科學家們對某種疾病或異常細胞的特定化學途徑(或致病原因)的基礎研究。這類基礎研究是在世界各地的學術性實驗室和研究機構中進行的,部分由製藥行業提供經費。
科研人員必須首先確定一個靶標。藥物靶標是身體中的分子結構,可與潛在的藥物化合物相互作用,產生治療或預防疾病的臨床效果。研究人員在細胞、組織和動物模型中進行研究以確定靶標是否受藥物影響,這個工作被稱為“靶標鑒別”。靶標鑒別是幫助科學家避免看起很有希望的研究走入“死胡同”的一個關鍵步驟。成功的靶標驗證可幫助科學家在實驗室確定最有希望的方法,以及提高研發效率。
Step2 藥物發現
階段目標 先導化合物選擇和優化
科研人員在深入了解疾病機製並確定可能的靶標之後,可將尋找的化合物範圍縮小為一個先導化合物,它可能作用於該靶標,並有可能成為一種新藥。
尋找先導化合物有多種途徑。
源自於自然界的化合物有悠久曆史,例如在土壤和黴變植物中發現的細菌誕生了一些重要的新藥,大自然也提供許多有用的物質。研究人員采用了許多途徑發現自然界來源新藥。
化學的進步使得科學家能夠通過使用複雜的計算機建模技術來預測可能奏效的分子類型,從而“從零開始”創建分子。高通量篩選是目前最常用的方法之一。機器人和計算機技術的進步使得科研人員能夠高效測試成千上萬個針對靶標的化合物,以識別出可能有前途的先導化合物分子。
此外,利用生物技術工具,科學家可以通過基因工程生物係統來產生對抗疾病的生物分子。
先導化合物分子必須經過檢測、分析,量化測試其與生物學靶標的相互作用,探索化合物改變生物靶標的行為方式。
檢測的化合物可保存在大型化合物庫中。大多數化合物通過化學合成技術獲得,天然來源如取自植物、真菌、細菌和海洋生物的產品也可被整合到化合物庫中。
通過測試成千上萬個相關化合物,確定哪一個具有更高的有效生物活性、更少的毒性以及更優良的藥理學性質。這些數據有助於研究人員篩選先導化合物,隨後還要經過一係列測試,對其安全性進行早期評估。
一個新藥必須符合以下五個條件:1.能被吸收進入血液中;2.能分布至身體的適當部位;3.能有效地被代謝;4.能成功地從身體排出體外;5.證明沒有毒性。
因此,科學家需要測試每種先導化合物的吸收、分布、代謝、排泄和毒理學(ADME/Tox)特性(藥代動力學)。這些研究幫助科研人員初步篩選先導化合物。ADME/Tox研究在活細胞、動物和計算模型中進行。
下一步,要對初篩後幸存下來的先導化合物進行優化或改良,使它們更有效和更安全。采用計算機模擬先導化合物的結構,以及與目標蛋白質鏈接,這種基於結構的設計方法被稱為“計算機模擬”(in silico modeling)。結構信息技術使科學家能以更合理的方式修飾所選的分子或化合物。
在改變化合物的結構時,科學家可以通過設計使其具有所需性質,例如減少或防止化合物與身體中的其他化學途徑相互作用,從而減少出現副作用的可能性。還需要對初始先導化合物數百種不同變體或“類似物”進行測試,實現優化。先導化合物經過優化便可獲得具有良好生物和化學特性的候選藥物,隨後可對這些優化的先導化合物進行臨床前試驗。
Step3 臨床前試驗
階段目標 確定可否在人體內研究
臨床前試驗的主要目的是確定一個先導化合物可否在人體內進行研究。
這個目標的首要任務是評估其可能造成的嚴重危害。臨床前研究可提供先導化合物劑量和毒性水平的詳細信息,確定其是否致癌、致突變或致畸等。臨床前研究使用動物,通常無須非常大的規模。
二是評估臨床試驗需要的初始劑量,並有助於確定安全性評估標準。後者包括臨床試驗期間應密切監測的患者體征和症狀等。臨床前研究提供的藥理學概況可幫助科研人員規劃最初的藥物生產過程和藥物劑型,以用於稍後進行的人體測試。
三是設定早期階段被評估藥物的化學品質、純度和穩定性,以及確定產品質量和純度重現性的生產製造過程規範。
在這個階段,還要確定如何保障有足夠量的藥物用於臨床試驗,因為臨床前階段使用的小規模生產工藝技術有可能不易轉化為更大規模生產。如果候選藥物被批準用於一般患者群體,那麽就需要考慮生產擴大規模的相關問題。
臨床前研究有助於科研人員設計下一階段的研究,即在人體進行的Ⅰ期臨床研究。一旦完成臨床前測試,在開始人體測試之前必需向FDA提交研究性新藥(IND)申請。此類申請文件應包括臨床前研究結果、候選藥物的分子結構、如何在人體內發揮作用的詳細信息、臨床前研究出現的潛在副作用,以及製造工藝信息。IND還應提供詳細的臨床試驗計劃,說明如何、在哪裏以及由誰進行研究。如果IND申請獲得批準,就可以開始臨床試驗。
Step4 臨床試驗(Ⅰ期~Ⅲ期臨床)
階段目標 在人體中評估安全性和功效
臨床前研究使用動物模型回答了藥物安全性的基本問題,但不能代表候選藥物與人體相互作用的方式。而臨床研究是指在人類受試者中進行的研究或試驗。科研人員從IND出發,設計臨床研究,以順利完成不同階段的每項研究任務。臨床試驗需要循序漸進,遵循從早期小規模的Ⅰ期研究到晚期大規模Ⅲ期研究的典型次序。
Ⅰ期臨床
階段目標:初始安全性測試
Ⅰ期試驗是首次在一組健康人類誌願者中進行藥物測試。研究的重點是評估實驗性候選藥物(1~5個)的安全性,確定安全劑量範圍,確定副作用。如果在疾病患者中開展研究,則需要檢測有效性的早期證據。
Ⅰ期臨床設計Tips
受試者數:20~100名健康誌願者或患有疾病/症狀者
時間:幾個月
目的:安全性、耐受性、劑量和藥代動力學
內容:安全性和耐受性研究(單次遞增劑量/多次遞增劑量)、藥物相互作用、食物效應、年齡和性別的影響
通過率:約70%進入下一階段
Ⅱ期臨床(包括ⅡA和ⅡB)
階段目標:小規模患者中評估安全性和功效
在Ⅱ期臨床試驗中,被研究的實驗性藥物首次在疾病患者或該藥針對的症狀患者中進行檢測。這有助於確定正確的劑量、常見的短期副作用,以及用於較大規模臨床試驗的最佳方案。Ⅱ期臨床可分為ⅡA和ⅡB試驗。在ⅡA階段,目標是進行初步的劑量範圍探索,並獲得初始的概念證據(POC)。POC需證明藥物起到了所需發揮的作用。ⅡA和ⅡB一起被稱為“探索性開發”,而Ⅱ期臨床和Ⅲ期臨床則為確定性開發階段。ⅡB期試驗規模較大,可能使用對照藥物和更廣泛的劑量範圍進行比對,來獲得更穩定的POC。
Ⅱ期臨床設計Tips
受試者數:數百(100~500)名疾病/症狀患者
時間:幾個月~2年
目的:功效、副作用、藥效學或生物學活性
內容:(ⅡA):概念驗證、功效、機製、劑量範圍探索;(ⅡB):確定治療劑量範圍下的結果
通過率:約33%進入下一階段
Ⅲ期臨床
階段目標:較大患者群體中證明安全性和有效性
Ⅲ期臨床試驗旨在證明實驗性藥物在患有該疾病的大型目標患者群體中的益處,目的是證實療效、監測副作用,有時將實驗性藥物與常用治療藥進行比較。科研人員還需利用這些臨床試驗收集藥物總體風險-效益關係的進一步信息,並為其成功獲準和撰寫說明書提供充分的依據。
Ⅲ期臨床研究在數百至數千目標疾病或症狀患者人群中進行,通常會在世界各地的多個臨床中心(即多中心研究)開展數年。這些研究提供滿足監管機構所需的證據,證明該藥物符合批準和上市的各項法律要求。
Ⅲ期臨床設計Tips
受試者數:300~3000名有目標疾病或症狀的誌願者
時間:1~4年
目的:有效性和不良反應監測
內容:注冊所需的關鍵性研究(與安慰劑和/或對照藥品比較)、長期安全性研究、上市後研究承諾
通過率:25%~30%進入下一階段
Step5 監管機構審查和批準
Step6 生產準備
(同時進行)
除了規劃臨床試驗,製藥公司的科技人員需同時開展製造工藝研究,以確保能生產高品質的藥品提供試驗中使用,以及規劃批準後全麵生產。製藥公司還需匯集和準備注冊審批所需的各種申請文件。
為了能夠盡快向嚴重疾病患者提供有效藥物,或某些治療領域醫療需求未得到滿足的情況下,FDA製度性地設立幾項加速新藥開發和審批的程序:
綠色通道(Fast Track):用於重症治療的藥物,且已有臨床前及臨床數據證明其有可能滿足現有療法無法滿足的醫療需求;或符合規定的感染類疾病治療的藥物。
突破性療法(Breakthrough Therapy):用於重症治療的藥物,且早期臨床數據表明,臨床療效可能明顯優於現有療法。
加速批準(Accelerated Approval):用於重症治療的藥物,且在某些方麵優於現有療法,並有一個替代性的臨床指標,據此能夠比較合理地預測出藥物的臨床有效性,或有一個早期臨床指標,據此能夠比較合理地預測出藥物對不可逆轉的病態或死亡或其他臨床指標的有效性(例如達到替代終點,該終點可以預測臨床獲益性)。
優先評審(Priority Review):對能顯著改善治療、診斷或預防嚴重疾病的安全性或有效性的候選藥物,優先評審以加速審批過程。
有的項目可同時獲得多種資格認定,尤其是突破性治療藥物。
Step7 上市後監察
Ⅳ期臨床試驗階段目標 上市銷售和安全監測
Ⅳ期臨床試驗是在藥品獲得FDA批準後進行的研究,研究結果可能被用於藥品營銷。有的是FDA要求,有的是製藥公司自願進行批準後研究,以獲得藥品長期安全性和有效性的其他信息,包括其風險、效益和最佳應用方式。
批準後的臨床試驗也可設計成對特定患者人群(例如對兒科患者)、新的給藥方式(例如定時釋放膠囊),或用於新適應症。由於獲準後臨床試驗旨在為取得FDA批準新用途或新給藥方式(製劑)提供依據,因此必須符合與初次獲準時所進行的Ⅲ期臨床試驗相同的標準。
Ⅳ期臨床設計Tips
研究對象:數千名患有目標疾病/症狀的誌願者
目的:安全性和有效性
結語
從發現小的化合物開始,證明其對目標疾病的有效性,隨後進行檢測,到臨床試驗證明既安全又有效,直至最終開發成一種新藥,通常可能需要10年或更長時間,並且耗費高昂。新藥發現和開發各階段的試驗目標、受試者數量、花費時間、費用占比見表1。在此過程中,許多潛在的藥物在發現和開發過程中被淘汰。

但是,這個艱難曆程為可能威脅生命和備受疾病折磨的患者帶來生的希望或改善生活質量。如果學術界、產業界和監管人員避開喧囂,以患者為中心,那麽這樣的艱難曆程都是值得的。
我國也有相應的藥物開發和評審體係,尤其是藥品創新程序需要完善,本文的內容或許可以作為借鑒。




























 相關新聞
相關新聞

 關於我們
關於我們










