9月11日,CFDA發布《已上市中藥生產工藝變更研究技術指導原則》(簡稱“正式稿”,下同)的通告。在中藥正式稿發布前, CFDA於8月29日發布了《已上市化學藥品生產工藝變更研究技術指導原則》。9月7日,CDE開始網上征求《生物製品上市後變更研究技術指導原則》意見。
如此,三類藥品的工藝變更研究技術指導原則有望在2017年全部發布完畢。而隨著工藝變更指導原則全部發布,CFDA飛檢就有了理論依據。工藝核對的飛檢,對於企業來說關乎生存,因此各生產企業應盡快開展工藝變更研究。
工藝變更飛檢已經開始
2016年8月9日,CFDA公開征求《關於開展藥品生產工藝核對工作的公告(征求意見稿)》的意見,原定於2016年9月10日修改意見征集完畢,藥品生產企業於2016年10月1日前完成自查並將自查情況報所在地省級食品藥品監管部門。省級食品藥品監管部門應對企業自查情況進行匯總,填寫自查情況匯總表並於2016年11月1日前上報食品藥品監管總局。
按征求意見稿,藥品生產企業應於2017年6月30日前完成在產品種生產工藝的研究驗證、實際生產工藝與批準生產工藝不一致的提交補充申請等相關工作,其他暫不生產品種應於2017年12月31日前完成上述工作;未按時完成的,2017年6月30日停止生產。
此外,2016年11月1日起,CFDA會組織專家對藥品生產企業針對生產工藝開展飛檢。如表1所示,2016年12月CFDA在飛檢時就已經開始檢查工藝了。
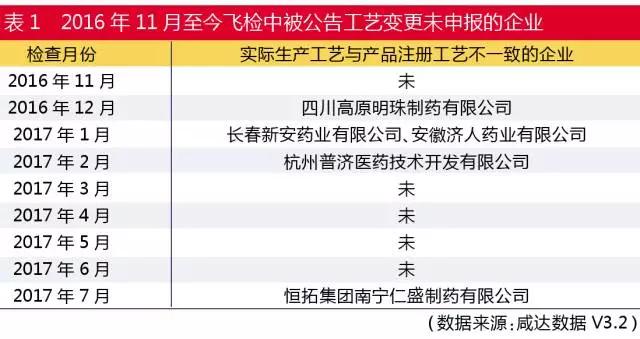
以2016年12月9-11日接受飛檢的四川高原明珠製藥有限公司為例,CFDA公告該企業複方板藍根顆粒生產批量由5萬袋變更為48萬袋,提取設備變更前使用1個提取罐,目前使用4個提取罐生產,未進行變更控製,且未對變更的批量進行風險評估和工藝驗證。由此可見,工藝變更已經是CFDA的飛檢項目內容之一了。
中藥“正式稿”九個不同細節
2017年3月,CDE網站發布關於《已上市中藥生產工藝變更研究技術指導原則》征求意見(簡稱“征求意見稿”,下同)的通知。鹹達數據V3.2對“征求意見稿”與剛剛發布的“正式稿”進行比較,發現主要有以下9個細節變更。
1.申請人對已上市中藥擬變更生產工藝開展研究,“征求意見稿”中的“並向藥品監督管理部門提出補充申請”字眼被刪去。
2.認可工藝變更往更好的質量方向提升。“正式稿”要求“無論何種類別的變更,都不應對藥品的安全性、有效性發生負麵影響”,“征求意見稿”原文是“不允許安全性、有效性發生明顯改變”。
3.中藥生產工藝變更研究的質量方法舉例,除了“征求意見稿”提到的指紋圖譜(特征圖譜)和溶出度檢查以外,“正式稿”增加生物活性檢測。中藥生產工藝變更研究及申報資料要求的Ⅰ類變更的“質量對比研究”中,“正式稿”也增加了生物活性檢測。
4.研究驗證樣品從“中試以上規模”改為“能夠代表生產實際情況的”,中藥生產工藝變更研究及申報資料要求的Ⅰ類變更中對“中試生產數據”的要求也從“提供3批穩定的中試研究數據”改為“提供3批能夠代表生產實際情況的中試研究數據”。
這基本與目前注冊法規大趨勢一致,例如新化學藥分類注冊資料要求生產工藝以目前生產的最大批量。
5.藥材前處理變更分類的Ⅰ類變更刪除了“不含熱敏性、揮發性成分的藥材或原粉增加瞬間高溫滅菌處理步驟”。
6.提取純化的變更分類的Ⅰ類變更中的“藥液靜置、過濾”改為離心;藥液中的“固形物含量不變”改為“藥液靜置”,過濾改為離心(或離心改為藥液靜置、過濾);藥液中的“固形物及指標成份含量不變”,“正式稿”對指標含量的變化有要求。
成型工藝變更分類的Ⅰ類變更,對於增加藥液普通過濾工序,或者變更藥液普通過濾的濾材材質、孔徑及過濾次數等工序,“正式稿”也新增要求“相關檢測及固形物、指標成份含量等不變”。
7.成型工藝變更分類的Ⅲ類變更,“正式稿”將“對藥物理化性質等有明顯影響的成型工藝方法的改變”改為“對藥物吸收利用等有明顯影響的成型工藝方法的改變”,更關注藥物動力學相關內容。
8.成型工藝變更分類的Ⅲ類變更中,“正式稿”刪去“緩釋/控釋製劑中緩釋材料種類或用量變更”。
中藥生產工藝變更研究及申報資料要求的Ⅲ類變更方麵,刪去“緩釋/控釋製劑變更輔料屬於Ⅲ類變更者,應提供藥代動力學研究資料”和“外用製劑變更輔料屬於Ⅲ類變更者,需要提供製劑非臨床安全性研究資料”兩項內容。
9.中藥生產工藝變更研究及申報資料要求的Ⅱ類變更方麵,臨床試驗或生物等效性研究比較資料正式稿刪去“其中臨床試驗研究進行病例數不少於100對的臨床試驗,用於多個病證的,每一個主要病證病例數不少於60對”的臨床試驗限製。
哪些企業進行工藝變更備案?
鹹達數據V3.2分析藥品注冊補充申請備案情況公示數據發現,截至2017年9月13日,2016年8月以後與工藝變更相關的補充申請共74條。74條基本都是不改變產品質量的生產工藝變更提出備案申請。
備案機構方麵,73條屬於重慶市食品藥品監督管理局,1條屬於陝西省食品藥品監督管理局。重慶的工藝變更補充申請備案數之所以排名第一,是因為2016年3月重慶發布了《重慶市食品藥品監督管理局關於加強藥品生產工藝及處方清理工作的補充通知》(渝食藥監藥生產[2016]14號)。藥品生產企業提出無質量差別的變更申請後,經重慶市藥品技術審評認證中心對申報資料審定,變更內容不影響產品內在質量,才能予以備案。
生產企業方麵肯定也是重慶的企業包攬前三。福安藥業(含慶餘堂製藥)受理號數最多,有37個;其次是天聖製藥,32個,重慶賽維藥業有限公司以2個排第三。
藥品分類方麵,化學藥品72條工藝變更,生物製品和中藥各1條。化學藥品變更的產品以抗生素的注射液和原料藥為主。生物製品變更工藝的是酪酸梭菌活菌膠囊,中藥則是薏辛除濕止痛膠囊。
小結<<<
從重慶的數據可推測,各企業對工藝變更的申報主要為“不改變產品質量的生產工藝變的備案補充申請”,預計一些工藝技術較成熟的注射劑會較容易獲批備案。
2016年臨床自查核查以來,大批量的申報被撤回,個別企業對於工藝核對的申報有點草木皆兵,生怕被秋後算賬。畢竟臨床自查核查對企業來說,屬於新產品開發受挫;一致性評價則可視為產品在醫療機構采購渠道受限;工藝核對對於企業來說可謂生存之戰,一旦飛檢不能通過,原有的產品可能被停產做工藝變更研究,GMP資質還有被取消的風險,企業豈能不重視?
隨著工藝變更指導原則全部發布,為CFDA飛檢提供了理論依據。各生產企業要盡快開展工藝變更的研究了。




























 相關新聞
相關新聞

 關於我們
關於我們










