口服固體製劑一致性評價申報資料要求“試行稿”的八個亮點 (附與“征求意見稿”的對比)
8月17日晚,CFDA發出《化學藥品仿製藥口服固體製劑質量和療效一致性評價申報資料要求(試行)》(簡稱“試行稿”,下同)。對比《化學藥品仿製藥口服固體製劑一致性評價申報資料要求(征求意見稿)》(簡稱“征求意見稿”,下同),“試行稿”主要有以下8個方麵的變化。
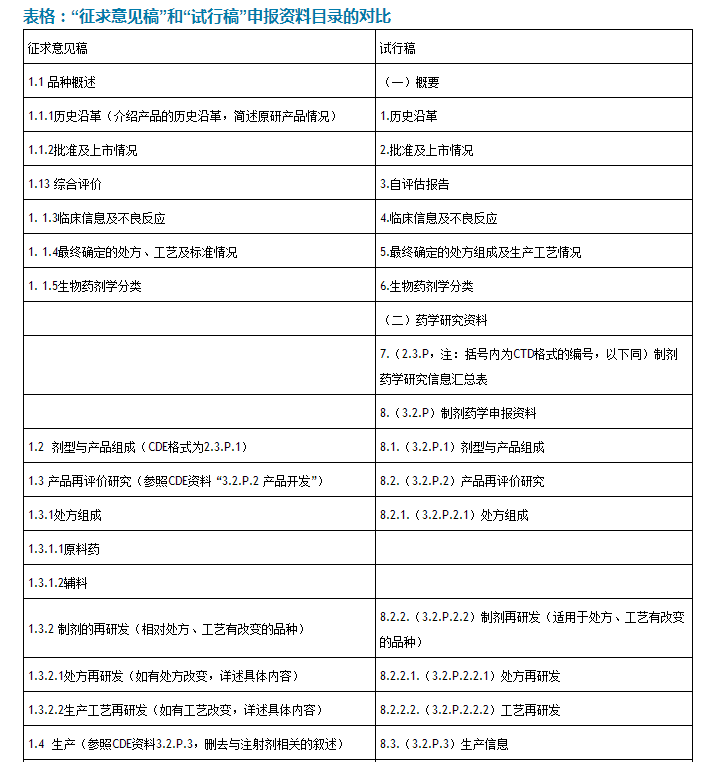
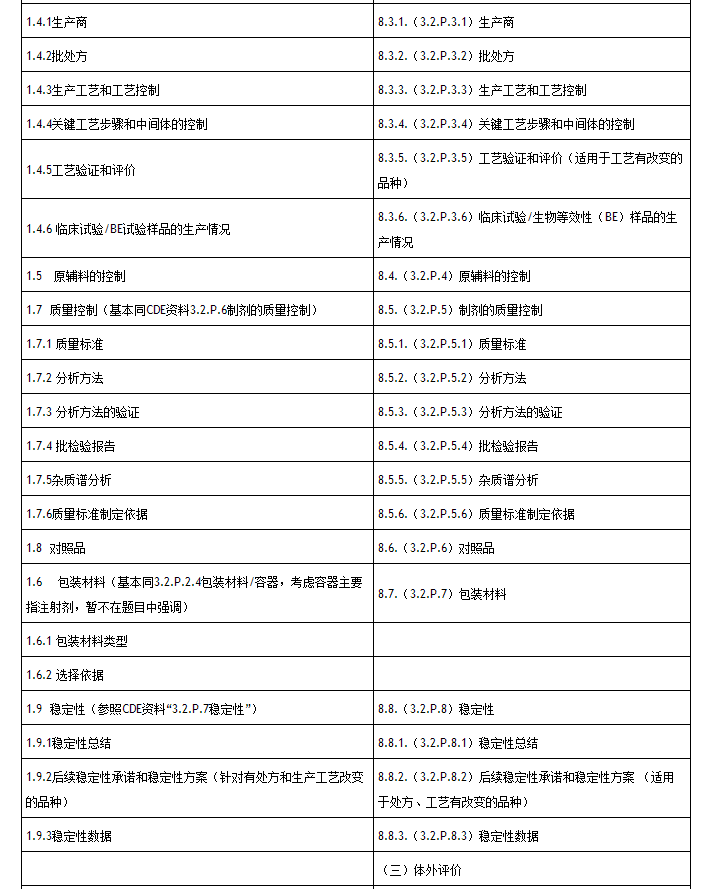
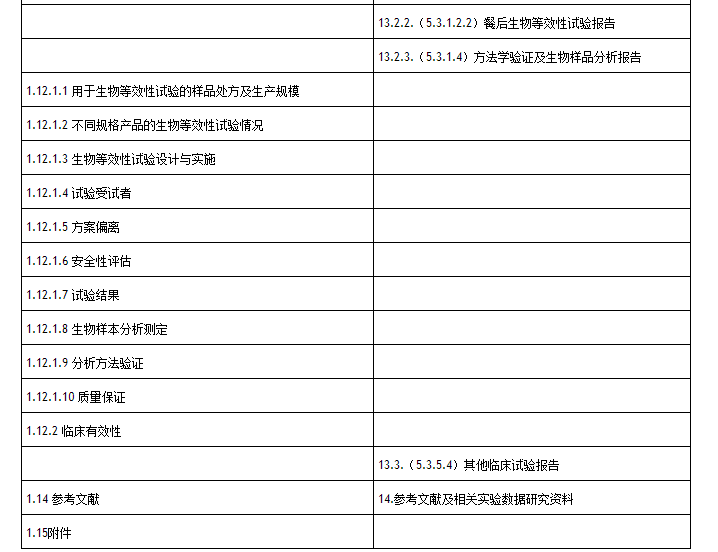
對比“征求意見稿”和“試行稿”的目錄,可以看出CFDA充分谘詢各家意見後對一些細節進行修訂,並且參考了目前已公告的內容,特別是化學藥品新注冊分類的申報資料要求,進行了統一規範,以免文件之間出現衝突。
1、對原研產品信息提供相對放寬
從文件多處可見原研產品信息提供要求相對合理。
如曆史沿革不再要求寫同品種原研產品立題的合理性分析。
允許企業通過文獻或專利信息資料作為原研產品和參比製劑的組成以及生產工藝概述(盡可能了解其特殊的、關鍵的工藝技術,例如濕或幹法製粒、有無預處理等)、輔料與內包材情況證據。
企業亦可通過提供原研藥或參比製劑的質量概況在內的相關研究資料或文獻資料來說明原研藥或參比製劑的質量概況(QTPP)。
此外,如果原研產品信息真的不能提供,企業可作相應說明應對,如僅涉及工藝變化、未涉及處方變化的品種,在無法提供原研產品或參比製劑具體信息可作相應說明。
2、一致性評價也要按CTD格式
“試行稿”要求藥學研究資料按照CTD規定的格式和撰寫要求,提供製劑藥學研究的主要信息綜述資料。
這意味著化學藥品的各種申報資料基本都要按CTD格式要求申報了。
在“試行稿”中,對於仿製藥一致性評價的申報,除製劑藥學研究信息匯總需要CTD格式外,其他部分暫無此要求。
3、“自評估報告”關注科學性、完整性和真實性
“自評估報告”是根據產品該次一致性評價具體內容的相關研究所進行的全麵論述。需結合每項研究內容,對處方工藝研究、關鍵步驟和中間體的控製、原輔料、包裝材料進行分析,重點針對與參比製劑一致性相關的質量研究情況、體內評價研究結果等的分析,提出對該品種與參比製劑質量和療效一致性的綜合評價結果。
實際上,“自評估報告”部分為“征求意見稿”的“1.13綜合評價”內容提前。
“自評估報告”還要求申請人保證該品種研發過程及結果和申報資料的科學性、完整性和真實性。
4、生物等效性試驗報告包括空腹試驗和餐後試驗
“試行稿”目錄要求生物等效性試驗報告包括空腹試驗和餐後試驗,具體內容和要求參考《關於發布化學藥品新注冊分類申報資料要求(試行)的通告》(國家食品藥品監督管理總局通告2016年第80號)附件第二部分注冊分類4、5.2類申報資料要求(試行)附件5製劑臨床試驗資料。
而“征求意見稿”則僅僅要求說明是否進行了空腹和餐後生物等效性試驗。口服固體製劑的人體生物等效性試驗一般應進行空腹給藥及餐後給藥的等效性研究。若僅進行一項研究(空腹或餐後等效性試驗),應提供免除另一項研究的充分理由。
5、不同溶出儀之間結果差異作為體外溶出試驗方法學驗證
在“征求意見稿”中,體外溶出試驗方法的耐用性主要是針對色譜係統包括色譜柱、流動相的耐用性。
在“試行稿”中,則將不同溶出儀之間的結果差異考察作為建立體外溶出試驗方法的耐用性溶出量檢測方法的方法學驗證結果考察。
此外,體外溶出試驗方法學驗證的溶液穩定性中,“試行稿”還要求不同溶出介質應分別考察主藥成分的穩定性。
6、溶出曲線穩定性考察適用於理化性質不穩定品種
“試行稿”對溶出曲線穩定性考察的適用對象有了限製,主要適用於理化性質不穩定品種。對有文獻報道或者研究資料表明有光照、高濕、高溫、氧化等條件下不穩定的品種,建議考察參比製劑溶出曲線穩定性,為實驗室複核結果的重複性提供支持。
如參比製劑溶出曲線結果與零時間點(t0)的溶出曲線比較變化較大,應根據具體情況調整試驗方法,並將相應的試驗方法和注意事項與複核單位溝通,保證複核結果的一致。
申報資料要求提交至少3個月加速試驗和3個月長期試驗的溶出曲線數據以及結論,以表格形式整理。“試行稿”建議申報單位在長期試驗條件下同時將參比製劑以及仿製製劑留樣保存,直至藥檢機構複核試驗結束或至有效期止。
7、關注產品的臨床使用情況
CFDA越來越關注產品的安全性和臨床療效,如輔料的關注點不再僅限於是否適合所用的給藥途徑,“試行稿”還關注其每日安全用量。
臨床信息及不良反應部分,除了“征求意見稿”所要求的搜集主要不良反應信息,“試行稿”還要求提供再評價產品的臨床使用情況。
8、生物藥劑學分類需要參照《人體生物等效性試驗豁免技術指導原則》
生物藥劑學分類除了“征求意見稿”要求的列表說明不同文獻報道的生物藥劑學分類,“試行稿”要求必要時可參照《人體生物等效性試驗豁免技術指導原則》。





























 相關新聞
相關新聞

 關於我們
關於我們










