





Emai:marketing@yakkaa.com
業務谘詢專線:400-780-8018
Tel: +1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email: marketing@medicilon.com
地址:上海市浦東新區川大路585號
郵編:201299
電話:+86 (21) 5859-1500(總機)
傳真:+86 (21) 5859-6369







© 2023 上海hjc黄金城生物醫藥股份有限公司 保留所有權利 滬ICP備10216606號-3
滬公網安備 31011502018888號 | 網站地圖











業務谘詢
中國:
Email: marketing@yakkaa.com
業務谘詢專線:400-780-8018
(僅限服務谘詢,其他事宜請撥打川沙總部電話)
川沙總部電話: +86 (21) 5859-1500
海外:
+1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email:marketing@medicilon.com




根據藥品注冊管理辦法附件二的規定,仿製藥即是已有國家藥品標準的原料藥或者製劑,該類藥物國內已批準生產或上市銷售,經過國內外廣泛使用,其安全性、有效性已經得到較充分證實。
如今的新法規對仿製藥提出了新的要求,主要是以下幾點:
1、規範對被仿製藥品的選擇原則,即參比製劑的選擇問題。
2、增加批準前生產現場的檢查。
3、按照CTD格式要求提供申報資料,使申報規範,統一。
4、強調了對比研究,是判斷兩者質量是否一致的重要方法之一。
5、強化了工藝驗證,目的是確保大生產時能始終如一地按照申報工藝生產出質量恒定的產品。
6、提出了晶型的要求,晶型的不同,溶解度和穩定性不同。
分析上述新要求和參考指導原則,從而得出結論:
仿製藥研發的目的是做到規模化生產,強調本地化,以實現“替代性”。要求是做到“同”。方法為對比研究。
1.安全性“同”:
對於安全性,口服固體製劑控製的主要為有關物質,而液體製劑除控製有關物質外,還需對防腐劑、氧化劑等對人體有影響的物質進行控製。因此,必須要將防腐劑含量測定定入質量標準。
研究的內容:靜態上應包括雜質譜的對比,單個雜質的對比,雜質總量的對比。動態上的對比為影響因素試驗、加速試驗的對比,即穩定性對比研究。
2.有效性“同”:
對於口服固體製劑,口服混懸劑(包括幹混懸劑),溶出曲線是主要的控製指標[1];對於口服溶液劑,防腐劑、矯味劑、氧化劑、增溶劑及穩定劑的選用非常重要,控製點為口感、滲透壓、PH及有無絮凝現象;對於局部用製劑(如鼻噴霧劑),粒度分布、滲透壓及黏度是主要控製指標。
研究的內容:分別進行溶出曲線對比;粒度分布對比;滲透壓及黏度對比。
3.晶型:
晶型的不同,藥物的溶解度及穩定性有可能不相同,從而導致生物利用度不盡相同。而某個藥物的晶型,文獻資料很少;製劑中原料的晶型測定有一定的難度;在做成製劑的過程中,又不能保證晶型不產生變化。
但是,鑒於仿製藥研究的特點,溶解度方麵可通過溶出曲線對比來說明;穩定性方麵可通過影響因素試驗和加速試驗的對比來說明。
hjc黄金城生物醫藥在仿製藥質量一致性評價方麵,具有豐富的仿製藥研發經驗,可以提供:參比製劑和仿製藥的質量全麵比對、處方工藝二次開發服務、動物BE服務等服務;報告模板和SOPs方麵,能夠符合客戶要求完成相應分析條款,提供準確及合規的文件,提供可追溯性完整性等高質量原始數據,符合NMPA的要求。
(從立項到申報,時間為10—12個月)
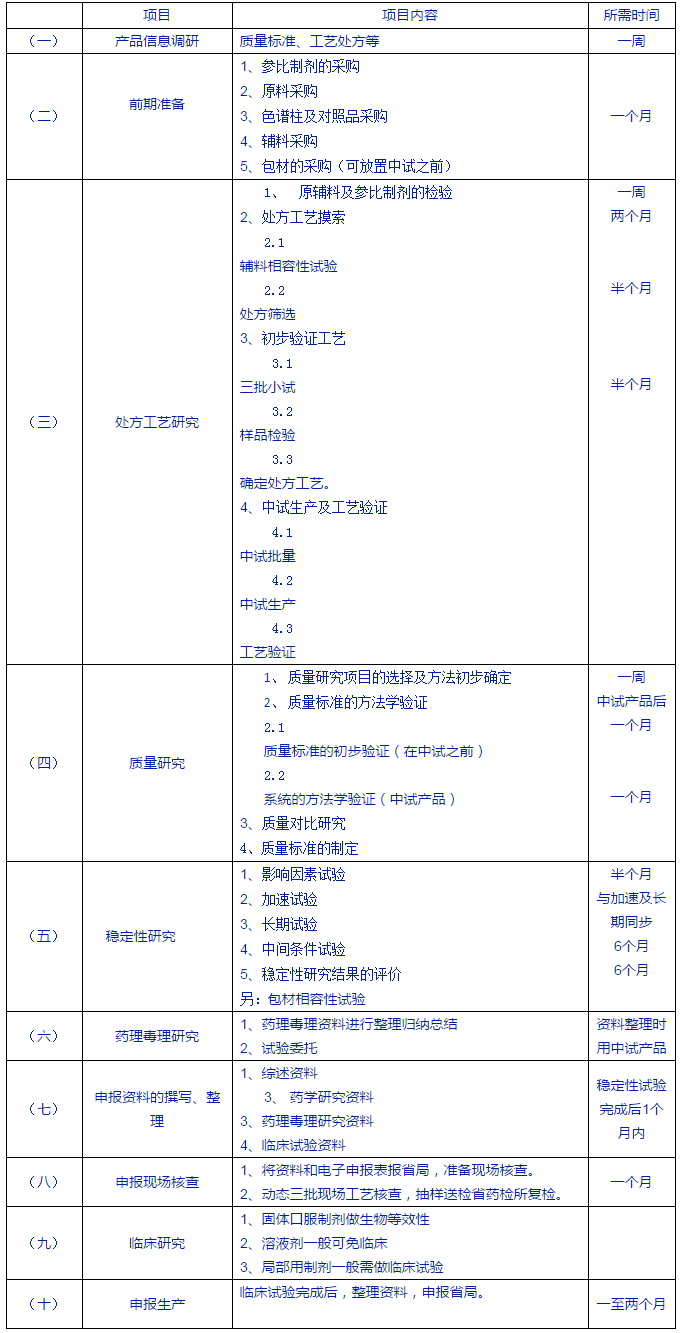
(一) 產品信息調研
(約一周完成)
是否有合法原料提供;臨床資料、不良反應資料及產品說明書等相關資料;國內及進口製劑劑型及規格;產品質量標準(原研標準、國內首仿標準、藥典標準);原研處方組成及工藝研究資料;藥品的穩定性資料;專利情況;生產注冊情況(產品原研廠家、國內生產申報廠家數情況);參比製劑來源等。
(二) 前期準備(約一個月完成):
1、 參比製劑的采購
1)首選已進口或本地化生產的原研產品;
2)如果無法獲得原研產品,可以采用質量優良的在發達國家上市的藥品,如在ICH成員國上市的同品種,即美國、歐盟或日本等國的同品種仿製產品。如果上述國家產品已經進口中國,可采用進口品。
3)如果無法獲得符合上述要求的對照品,則應在充分考慮立題合理性的前提下,采用多家國內上市的主流產品,進行深入的對比研究,所申報產品的質量應能達到其中最優產品的質量。
4)如果確實無法獲得符合要求的已上市對照品,在充分考慮立題合理性的前提下,應按照新藥研究的技術要求進行相應的研究。
2、 原料采購
可選用幾個廠家的小樣進行對比後,采購質量較好的(需提供原料廠家資質、發票、檢驗報告、標準、購銷合同及長期供貨協議等證明性文件)。
3、 色譜柱及對照品采購
在對原料質量標準、查詢到的製劑質量標準分析的基礎上,擬定標準草案。向原料供應廠家充分了解產品的色譜條件後,再對色譜柱及對照品進行采購。包括:色譜柱的型號,規格,生產廠家;對照品的種類(含異構體);對照品的規格;對照品的用途(UV或含測用);對照品采購量(注明價格)。
4、 輔料采購:
根據國內輔料應用情況,對原研藥的處方組成進行合理分析後確定輔料的采購(廠裏已有輔料不采購、需提供輔料廠家資質、發票、檢驗報告、標準、購銷合同等證明性文件)。
輔料選用標準:首選藥用級;無藥用級,口服製劑及局部用製劑可選用食用級。若也無食用級,考慮更換輔料。
5、 包材的采購:
在參比製劑購買以後,參考參比製劑的包裝材料,結合公司情況,擬定包材種類(廠裏已有包材不采購、需提供包材廠家資質、發票、檢驗報告、標準、購銷合同等證明性文件):包材的種類(口服或注射級);包材的規格(包裝規格);包材藥用標準(藥典標準或是注冊標準);采購量。此項工作可放緩。
(三) 處方工藝研究
1、 原輔料及參比製劑的檢驗
(約一周完成):
確定原輔料的合法來源;參照藥典標準或其他相關標準對原、輔料進行檢驗;出具檢驗報告書。
對參比製劑進行全麵的檢測,檢測項目應不僅限於擬定的製劑質量標準。如固體口服製劑應對參比製劑進行溶出曲線的測定,對PH值敏感的藥物製劑測定5%混懸液PH值;液體製劑應加測黏度、滲透壓及PH值等。檢驗結果匯總。
通過這一項目,可以基本了解製劑要達到的基本性能。
2、 處方工藝摸索
2.1 輔料相容性試驗
(1)口服固體製劑:
通過前期的信息調研中,得知原料性質比較穩定,對輔料及保存條件沒有太多要求時:
若能查到原研藥的處方組成,而擬定的輔料種類與原研藥一致的情況下,可不做此試驗;若在原研藥處方的基礎上,增加輔料種類,隻需做增加的輔料相容性試驗;若查不到處方組成,需做輔料相容性試驗。
具體做法:
選若幹種輔料,將輔料與主藥按一定比例混合,取一定量,參照藥物穩定性指導原則中影響因素的實驗方法,在高溫60度、強光、高濕(RH75%、RH92.5%)試驗。分別於0天、5天、10天取樣。重點考察性狀、含量、有關物質等,必要時,可用原料藥和輔料分別做平行對照實驗,以判別是原料藥本身的變化還是輔料的影響。
對原料性質完全不了解,或通過信息調研,得知原料性質不穩定,對輔料及保存條件有特殊要求時:
即使查到原研藥的處方組成,仍建議做一輔料相容性試驗,因為原輔料生產廠家不同,穩定性也不同,雜質種類也可能有差別。在這種情況下做法可以相對簡單一些。
例:經過查找原研藥的處方,所用輔料為:XXX XXX XXX,在此基礎上,將原料和所有輔料按一定常規用量混合,用原料及空白輔料做平行對照,在高溫60度、強光、高濕(RH75%、RH92.5%)試驗。分別於0天、5天、10天取樣。重點考察性狀、含量、有關物質等。
(2)液體製劑:液體製劑可不進行此項試驗。
2.2 處方篩選
(一般采用單因素試驗的方法,必要時候可采用正交法)
通過上述的輔料相容性試驗,對主藥的穩定性有了基本的認識。
(1)固體口服製劑:
①先按照輔料的常規用量和常規工藝,以製劑基本性能(如口服固體製劑顆粒的可壓性、流動性及藥片的硬度、脆碎度、水分等)為指標進行初步篩選。
②選出兩到三個基本性能合格的處方樣品,進行溶出度曲線測定,與原研製劑進行比較,找出差距,調節輔料的用量,使溶出曲線達到一致。
③確認兩個或三個最佳處方工藝,分別作出小樣,和原研產品對比進行影響因素研究,研究項目根據輔料相容性試驗的結果來確定,基本上選用主藥敏感的因素即可!不要求全部因素都做。
④初步確定處方工藝。
(2)液體製劑:
①根據參比製劑的基本性能如黏度、口感、滲透壓、PH值等來進行輔料的用量選擇。
②對於防腐劑的用量,若可以在原研的說明書或質量標準中查詢到用量,可直接參照原研藥的用量。若沒有文獻資料,需做一抑菌性試驗,選用最低有效量。
具體做法:首先參考該防腐劑的常規用量,然後設定三至四個濃度,可以用等比的濃度。例如X的常規用量為0.2%—1.0%,可選用0.1%、0.2%、0.4%、0.8%四個濃度來進行試驗。試驗操作和判定標準參照中國藥典附錄。
③選出一至兩個基本性能合格的處方樣品,與原研進行影響因素試驗,實驗項目可適當簡化,如隻進行高溫60度這一條件即可,初步判定穩定性。
④初步確定處方工藝。
3、 初步驗證工藝
3.1 三批小試
用擬定的處方工藝放大生產三批,樣品規模片劑可為1000片/批,液體製劑可為500g/批。並填寫生產批記錄。
3.2 樣品檢驗
檢驗標準為參照原研、國內首仿或國內主流產品、藥典等擬訂的本品質量標準草案,草案應不低於被仿標準。產品合格,確定處方工藝;產品不合格,則重新進行處方工藝篩選。檢測結果符合質量標準,並與參比製劑一致,出具檢驗報告單。
3.3 確定處方工藝
在證實擬定處方工藝的可行性後,確定處方工藝。
4、 中試生產及工藝驗證
根據公司條件及相關指導原則,擬定中試生產和工藝驗證同時進行。因此,要求上一階段的工作必須紮實,檢測結果必須準確無誤。
4.1 中試批量:
根據法規要求和公司現狀,擬定每批批量為:口服固體製劑投料量為10公斤左右,根據此重量來折算萬片數;液體製劑也為10公斤左右。
法規要求:中試產品必須在GMP車間進行生產;所用設備應和將來大生產所用設備相同或原理及設備參數相同;批量為不少於以後大生產的1/10。
4.2 中試生產:
用確定的工藝在車間生產三批中試產品;填寫生產批記錄。
4.3 工藝驗證:
收集、評估整個工藝設計階段及生產全過程的數據資料,確立工藝能持續一致地生產出符合質量要求的產品的有科學依據的證據。
資料內容包括:工藝驗證的立項、方案、審批、報告、評價和建議、工藝驗證證書。
(四) 質量研究
在藥品研發中,質量研究是重點。參考指導原則,現將質量研究分成四個部分:質量研究項目的選擇及方法初步確定;質量標準的方法學驗證;質量對比研究;質量標準的製定。
1、 質量研究項目的選擇及方法初步確定
可稱為“質量標準草案的初步建立”,此項工作應在輔料相容性試驗之前完成。
1.1 遵循“就高不就低”的原則。結合所查詢的產品質量標準(原研標準、ChP、EP、BP、USP、JP等,如何查詢?)和藥典對具體劑型的要求,確定出質量標準草案。靜脈注射劑處方中加有抗氧劑、抑菌劑、穩定劑和增(助)溶劑等,眼用製劑處方中加有防腐劑等,應對相應的輔料進行定量研究。
對於國家藥品標準中收載的項目,首先應考慮選用標準中收載的檢測方法。
1.2若有關物質檢測方法多種並存時,建議初步對比研究來確定方法。如有雜質對照品,用雜質對照品來確認方法的可行性;如沒有雜質對照品,可做一強製降解試驗(需要特別注意的是降解程度為5%~10%左右,在此情況下判定物料平衡才有意義),來初步判定檢測方法的可行性。
判定標準:有雜質對照品時,係統適用性、分離度、有效檢出、精密度及重現性。
無雜質對照品時,係統適用性、降解雜質的有效檢出、物料平衡。
1.3 質量標準草案的初步建立。
2、 質量標準的方法學驗證
具體分為兩個方麵:方法的初步驗證和係統的方法學驗證。
2.1 質量標準的初步驗證(在中試之前完成)
①在配合處方工藝篩選檢驗時,就是質量標準初步驗證的過程。例如輔料相容性試驗、參比製劑與小試產品的對比檢驗、小試產品的影響因素試驗等,就可以對方法的可行性進行一個初步的判斷。
在這時,方法學研究偏重於驗證國家標準中的檢測方法和條件是否適用,重點考察方法的專屬性和準確度。如方法學研究結果顯示方法不適用,應首先分析原因,通過調整處方工藝等以使方法適用;在原因無法確認的一些極端條件下,才考慮建立新的檢測方法,但新方法首先要按照化學藥物質量控製研究相關指導原則進行研究,還需通過比較研究證實與原方法具有同等的控製程度。
因此,原則上不要更換已有的國家藥品標準的色譜條件。當分離度達不到時,可適當調整流動相的比例。
討論:在前期的處方篩選中,質量標準並沒有真正建立,最終確定的質量標準可能不一樣。這種情況下,認為處方篩選的數據仍然可以放入申報資料中,這和處方篩選的目的並不背離,同時也反映出質量研究的開展過程。
②出具三批小試樣品的檢驗報告書。
2.2 係統的方法學驗證
在初步驗證的基礎上,需對質量標準進行係統的方法學驗證。方法學驗證所用樣品應采用中試產品。
驗證項目(品種及劑型不同檢測項目不同):
性狀;鑒別(理化鑒別和光譜鑒別);一般檢查項(按中國藥典製劑通則);微生度檢測(需進行完整的方法學驗證試驗);溶出度,有些可以和含量一起驗證;
有關物質(需進行完整的方法學驗證試驗);含量測定(需進行完整的方法學驗證試驗)、衛生學方法學驗證。
其中重點是有關物質和含量的方法學驗證。
有關物質驗證的內容有:
係統適用性:
取樣品,按照有關物質供試品濃度配製溶液,進樣,記錄圖譜。理論板數應
符合規定,分離度應大於2.0或符合規定、拖尾因子應0.8-1.2或符合規定。
有已知雜質並且雜質對照品可獲得的:雜質對照品溶液連續進樣的峰麵積的相對標準差應不大於 2.0%,保留時間的相對標準差應不大於1.0%。另外,理論板數應符合規定,分離度應大於 2.0或符合規定、拖尾因子應0.8-1.2或符合規定。
專屬性:
空白溶劑幹擾試驗、空白輔料試驗、強製降解試驗(高溫、強光、強氧化、強酸、強堿等)、已知雜質定位試驗、峰純度檢查(二極管陣列檢測、質譜檢測)。
檢測限與定量限:
一般采用信噪比法。有已知雜質並且雜質對照品可得的,須用已知雜質對照品同時做。信噪比10:1,為定量限;信噪比為3:1,為檢測限。
線性範圍:
一般做5~7個濃度,如40%、60%、80%、100%、120%、150%(相對於自身對照濃度)的係列溶液。取該係列溶液進樣,記錄色譜圖,計算回歸方程。
若有已知雜質並且雜質對照品可得的,則取已知雜質對照品另作線性關係試
驗(以定量限為起始濃度點),供試品中已知雜質峰麵積,應在線性範圍內。
精密度:
包括重複性和中間精密度。重複性:6份供試品;中間精密度:不同時間、不同人員、不同儀器,6份供試品,與重複性試驗的6個數據一起進行比較。
溶液穩定性:
準確度:
一般以回收率試驗進行驗證。無已知雜質的,可不做。有已知雜質的,須做加樣回收試驗驗證準確度。
討論:有關物質的檢測中,要檢測的是雜質,而不是原料。因此,認為在有已知雜質對照品,並且在標準中對此雜質進行了單獨控製的時候,方法學驗證的內容應圍繞著已知雜質展開,不可以用原料來代替。當無已知雜質對照品時,才用原料代替(自身對照,如1%對照,0.5%對照,0.1%對照)。
在做強製降解試驗時,同時用空白輔料平行做強製降解試驗,特別是含有特殊輔料的,如防腐劑等在紫外有吸收的輔料。
關於強製降解試驗,不僅是方法學驗證的內容,而且是對產品的降解途徑、雜質譜及產品穩定性判定的過程。仿製藥相關指導原則要求仿製品與原研藥應比較雜質譜、雜質量及降解途徑。因此,建議把研試品與原研藥平行進行強製降解試驗(均約5~10%),這樣可以直觀比較兩者的降解途徑是否一致;降解物是否有差異;檢查方法對兩者的專屬性差別(由於擬定的方法多是原研藥的檢測方法)。平行對比破壞性試驗研究,是評價研製藥和被仿藥質量是否相同的重要手段。
關於強製降解試驗中物料平衡的問題:
①首先,降解強度為5%~10%左右。
②有雜質對照品時,計算出校正因子,將校正因子代入計算。
③做峰純度檢查(二極管陣列檢測),二極管陣列檢測的作用不僅是峰純度檢查,另外,還反映出雜質及主藥的總體紫外吸收情況。可以計算在不同波長處的物料平衡情況(按具體品種而定)。
①首先,降解強度為5%~10%左右。
④計算方法:通過與正常樣的總峰麵積對比。具體做法:建議取一定量的樣品溶解,作為母液,分別從母液中取一定量進行個因素的降解試驗,再與此母液做的正常樣進行對比,這樣做出的結果才可靠。
3)出具三批中試樣品檢驗報告書。
3、 質量對比研究
(采用中試產品)
質量對比研究是判斷仿製藥與被仿製藥質量 “一致性”或“等同性”的重要方法,可以全麵了解產品的質量特征,為仿製藥注冊標準的建立提供依據。
3.1 溶出曲線對比研究:
一般采用在四種溶出介質(如pH1.2、pH4.5、pH6.8和水)中的溶出曲線對比的方法,用f2因子法(f2>50)來比較原研藥和仿製品的曲線相似性。(溶出介質的選擇可參考謝沐風的文獻)
注意事項:
①用於比較的兩種製劑含量差值應在 5%以內。
②計算時所選取的時間點間隔無需相等,但兩製劑所取時間點必須一致;且計算時間點應不少於 3個;由於該計算結果有依賴於比較時間點個數的特性,故在溶出率
85%(緩釋80%以上)以上的時間點應不多於一個。溶出量應按累計溶出量來計算。
③除 0時外,第一選取時間點溶出結果的變異係數應不得過 20%,自第二時間點至最後時間點溶出結果的變異係數應不得過10%。如超出,應從儀器適用性或樣品均一性的角度考慮予以解決。
3.2 雜質的對比研究(此項內容可與方法學驗證同做):
對於有關物質檢查,由於原料藥製備工藝、製劑處方工藝的不同,仿製藥的雜質種類和被仿製藥可能不同,因此要求進行對比研究,分析仿製藥和被仿製藥中雜質的種類和含量情況。
①可通過強製降解試驗及影響因素試驗的對比研究,來比較仿製藥與原研藥的雜質種類、降解途徑及雜質大小。
②對於複方製劑來講,首先應對雜質進行歸屬。采用方法:分別做單個原料、空白輔料及製劑的強製降解試驗。
要求如下:
a. 研製產品中的雜質種類和含量實測值均不高於參比製劑。b. 如果某一雜質含量高於參比製劑,須不高於標準限度(一般為原研藥品的質量標準限度)。
c. 如果研製產品中含有參比製劑中未含有的新雜質,則建議首先通過改進處方工藝降低雜質含量或種類,使不超過鑒定限度,否則需進行雜質結構的鑒定,分析雜質的安全性並提供有關數據,必要時應進行相關的安全性試驗。
3.3 檢測方法的對比研究(與方法學驗證同時進行):
如果研究發現國家藥品標準中一些檢測方法不適用於研製產品,為進一步驗證是檢測方法存在問題,還是研製產品自身存在質量問題,可以采用被仿製藥進行對比研究。
4、 質量標準的製定
(結合對比研究結果、穩定性研究結果製定):
4.1 可在國家藥品標準的基礎上,參考國外藥典及參考文獻,增加必要的檢測項目。
4.2 檢測方法:如新建方法與國家藥品標準中收載方法相比無明顯優點時,因國家藥品標準已經過較長時間和多家單位的驗證,建議仍采用國家藥品標準中收載方法。
4.3 限度:有多種方法可參考時,限度的製定遵循“就高不就低”的原則。
4.4 分別製定貨架期標準及放行標準,即注冊標準和內控標準,寫入申報資料。
(五) 穩定性研究(中試產品)
1、 影響因素試驗:
取中試一批和參比製劑,除去內包裝,分散為單層置適宜的條件下進行。一般包括高溫(60或40度)、高濕(92.5%、75%)、光照試驗。分別於第5天和第10天取樣檢測,重點考察性狀、含量、有關物質,高濕試驗增加吸濕增重項。
以上為影響因素穩定性研究的一般要求。根據藥品的性質必要時可以設計其他試驗,如考察pH值、氧、低溫、凍融等因素對藥品穩定性的影響。對於需要溶解或者稀釋後使用的藥品,如注射用無菌粉末、溶液片劑、幹混懸劑等,還應考察臨床使用條件下的穩定性。
2、 加速試驗:
取擬上市包裝的三批樣品進行,建議在比長期試驗放置溫度至少高15℃的條件下進行。一般可選擇 40℃±2℃、RH75%±5%條件下進行 6 個月試驗。在試驗期間第 0、1、2、3、6個月末取樣檢測考察指標。如在 6 個月內供試品經檢測不符合質量標準要求或發生顯著變化,則應在中間條件 30℃±2℃、RH65%±5%同法進行 6個月試驗。具體溫度,可以參考原研藥的說明書中貯藏一項。
討論:關於穩定性試驗對比研究,法規要求研製產品的穩定性不得低於已上市產品的穩定性。在通過強製降解試驗和影響因素試驗的對比以後,對兩者的穩定性有了一定程度的了解。
①若產品本身比較穩定,參比製劑可隻取0月和加速6月時樣品來進行對比。
②若產品本身就不穩定,建議參比製劑和三批中試製劑同步進行對比。
3、 長期試驗:
長期試驗是在上市藥品規定的貯存條件下進行,目的是考察藥品在運輸、保存、使用過程中的穩定性,能直接地反映藥品穩定性特征,是確定有效期和貯存條件的最終依據。
取三批中試樣品在 25℃±2℃、RH60%±10%條件進行試驗,取樣時間點為0月、3月、6月、9月、12月、18月、24月、36月。
長期試驗時間的選擇應依據產品穩定性情況、與被仿製藥穩定性的比較情況、擬定有效期等綜合考慮。申請注冊時,一般應提供不少於6個月的長期穩定性研究資料。
4、 中間條件試驗
30℃±2℃、RH65%±5%(若長期試驗采用此條件,則可不再進行中間條件試驗)。
5、 穩定性研究結果的評價
根據穩定性研究的結果,結合原研藥的情況,確定包裝材料、貯藏及有效期。
另注意:包材相容性試驗:
包材的選擇參考原研製劑,最好與原研材質相同。對於口服固體製劑,用擬定的包裝做加速試驗及長期試驗即可;對於液體製劑和半固體製劑,需考察包材材料中的成分(尤其是添加劑成分)是否會滲出至藥品中,引起產品質量的變化(如力百汀塑化劑事件)。
就公司現有條件來講,這項是難點,隻有通過與包材商的溝通,請包材商進行自我控製。
備注:2013年2月6日,CDE發布了《化學藥物(原料藥和製劑)穩定性研究技術指導原則征求意見稿》。
(六) 藥理毒理研究
1)大多數仿製藥的研究隻需要提供藥理毒理文獻研究資料即可,這中情況下可以查閱國內外文獻數據,找到該藥物藥理毒理資料進行整理歸納總結。
2)局部用製劑應在GLP實驗室,根據品種需進行刺激性、過敏性、溶血性試驗。如丙酸氟替卡鬆鼻噴劑需進行刺激性試驗。
(七) 申報資料的撰寫、整理
(穩定性試驗完成後1個月內)
1、 綜述資料
1)藥品名稱。
2)證明性文件。需注意科技查新報告的委托查詢,一般在穩定性試驗中期委托查詢。
3)立題目的與依據(有國家局頒布的撰寫技術指導原則,可以在查詢資料後即可撰寫)。
4)對主要研究結果的總結及評價(有國家局頒布的撰寫技術指導原則,可以在進入穩定性考察後撰寫)。
5)藥品說明書、起草說明及相關參考文獻(根據原研藥的說明書)。
6)包裝、標簽設計樣稿。
2、 藥學研究資料(附件2為7—15項)
仿製藥(六類),必須按照國家局頒布的CTD格式來撰寫。其他類,推薦按CTD格式撰寫。
1) CTD格式申報主要研究信息匯總表
2) CTD格式申報資料撰寫要求
3、 藥理毒理研究資料
16)藥理毒理研究資料綜述(根據相關技術指導原則)
21)局部用製劑提交:過敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮膚、黏膜、肌肉等)刺激性等特殊安全性試驗資料和文獻資料。
4、 臨床試驗資料
28)國內外相關的臨床試驗資料綜述。
29)臨床試驗計劃及研究方案。
30)臨床研究者手冊。
31)知情同意書樣稿、倫理委員會批準件。
32)臨床試驗報告。
後兩項為報生產時所需資料。
(八) 申報臨床及申報現場核查
1)將資料連同電子申報表報省局,準備現場核查。
2)動態三批現場工藝核查,抽樣送檢省藥檢所複檢。
(九) 臨床研究
固體口服製劑做生物等效性;溶液劑一般可免臨床;局部用製劑一般需做臨床試驗。
(十) 申報生產
臨床試驗完成後,整理資料,申報省局。
目錄
一、綜述
二、仿製藥研發項目匯總
三、仿製藥的研發具體步驟:
(一)產品信息調研
(二)前期準備(約一個月完成):
1、參比製劑的采購
2、原料采購
3、色譜柱及對照品采購
4、輔料采購:
5、包材的采購:
(三)處方工藝研究
1、原輔料及參比製劑的檢驗
2、處方工藝摸索
3、初步驗證工藝
4、中試生產及工藝驗證
(四)質量研究
1、質量研究項目的選擇及方法初步確定
2、質量標準的方法學驗證
3、質量對比研究
4、質量標準的製定
(五)穩定性研究(中試產品)
(六)藥理毒理研究
(七)申報資料的撰寫、整理
(八)申報臨床及申報現場核查
(九)臨床研究
(十)申報生產
 相關新聞
相關新聞川沙總部
地址: 上海市浦東新區川大路585號
郵編: 201299
電話: +86 (21) 5859-1500(總機)
傳真: +86 (21) 5859-6369
海外:
Email: marketing@medicilon.com
Tel: +1(626)986-9880(U.S. - West Coast)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)


 關於我們
關於我們










